Ewing Sarcoma
Karen J. Marcus
Torunn Yock
Nancy J. Tarbell
James Ewing (1866-1943) first described the bone tumor that bears his name in 1921 (1). He observed that the malignancy was most common in teenagers, occurred in the metaphyseal and diaphyseal region of long bones or in the flat bones, was associated with pain and often fever, had a histologic appearance of highly vascular sheets of small round cells, and was quite sensitive to radiation (Fig. 9.1) (2).
Ewing was a leading authority on cancer. He became professor and chair of pathology at Cornell Medical School in New York City and went on to serve as director of the Memorial Hospital, playing a crucial role in the growth of what is now known as the Memorial Sloan-Kettering Cancer Center (2).
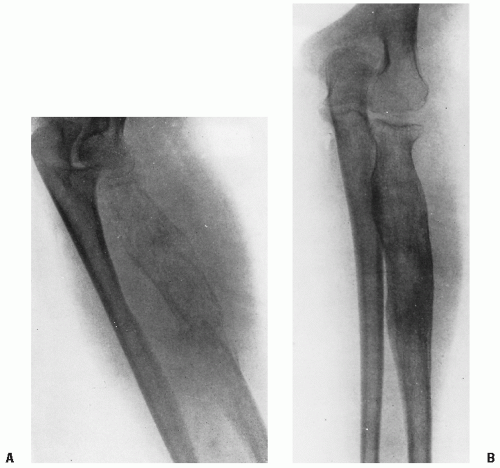 Figure 9.1 A: In the fourth edition of his textbook Neoplastic Diseases: A Textbook on Tumors (1940), James Ewing of the Memorial Hospital, New York, described a “diffuse endothelioma of radius, diffuse absorption without destruction of shaft, spontaneous fracture, wide invasion of muscle, duration 1 year.” Ewing wrote that “the first indication is for treatment by radiation in full doses, and over considerable periods. This recommendation is based on the reported cure of certain cases … by radiation alone, and on the clinical disappearance of the disease by variable periods in many more cases. The response to radiation also confirms the diagnosis. The danger of metastases occurring while this treatment is in progress is probably negligible, since the tumor tissue generally undergoes rapid liquefaction and necrosis.” B: Ewing illustrated the excellent response of the tumor to radiotherapy. |
Ewing sarcoma is the second most common childhood primary bone tumor. The tumor is slightly less common than osteosarcoma and represents 3% of pediatric cancers. Approximately 200 cases occur annually in the United States. Although it presents in the pubertal age range in 40% of patients, the age at diagnosis is more variable than that of osteosarcoma: 30% of cases occur in children younger than 10 years and 5% occur in young adults older than 20 years. Boys are affected more often than girls, at a ratio of 1.5-2:1. Age at
diagnosis parallels the earlier onset of puberty in girls, with a median time of onset 3 to 4 years younger than in boys. Ewing sarcoma is rare in children of Asian or African descent (3).
diagnosis parallels the earlier onset of puberty in girls, with a median time of onset 3 to 4 years younger than in boys. Ewing sarcoma is rare in children of Asian or African descent (3).
PATHOLOGY
Ewing sarcoma consists of monomorphic sheets of small, round malignant cells with hyperchromatic nuclei and little cytoplasm. There is a dearth of associated stroma. Cells usually are periodic acid-Schiff (PAS) positive, indicating the presence of glycogen granules. Glycogen in the tumor cells and the demonstration of the MIC2 gene product on the tumor cell membrane help identify Ewing sarcoma (4,5). The tumor cells are also uniformly vimentin positive and often cytokeratin positive, indicating origin from epithelial and neuronal elements (6).
Sarcomas are subdivided into two classes; first class is composed of tumors displaying complex karyotypic abnormalities with no distinct pattern, and the second class includes tumors associated with particular chromosomal translocations that result in specific fusion genes. The Ewing sarcoma family is in the second class of sarcomas. Ewing sarcoma and primitive neuroectodermal tumor (PNET) have been linked to the specific chromosomal abnormalities involving a reciprocal translocation between chromosomes 11 and 22: t(11;22) (q24;q12). This genetic rearrangement is detectable in 86% to 90% of tumors. Approximately 5% to 10% of Ewing sarcomas do not contain t(11;22) but instead have a translocation between chromosomes 21 and 22: t(21;22) (q21;q12). However, this alternate rearrangement has molecular consequences similar to those of t(11;22) (7).
The 11;22 translocation juxtaposes the EWS gene with the FLI gene, a member of the ETS transcription factor family. The 21;22 translocation juxtaposes the EWS gene to another ETS family member, ERG. There are structural similarities between these two translocations, suggesting that they bind to similar DNA target sites. The EWS-FLI1 fusion was the first sarcoma gene to be cloned (8). EWS-FLI can produce malignant transformation of some but not all cell lines. The EWS/FLI gene appears to act as a transcription factor. Its mechanism of action appears to be a modulation of transcription of target genes. Ewing sarcoma and PNET translocation seems to fall in a developing class of tumor-associated chromosomal translocations that form chimeric transcription factors (7). The formation of aberrant transcription factors is associated with many human malignancies. We may conclude that the probable mechanism of carcinogenesis in Ewing sarcoma is a translocation that produces an aberrant transcription factor. These factors can cause deregulation of pathways, resulting in tumorigenesis. Using retroviral systems to biologically screen cDNA from cells transformed by EWS/FLI1, platelet-derived growth factor-C (PDGF-C) has been identified as the target of EWS/ETS transcriptional deregulation (9). Therefore, Ewing sarcoma cytogenetics may help us to understand oncogenic mechanisms.
The EWS gene is a member of the ETS family of transcription factors defined by the ETS domain. The FLI1 gene appears to be involved in early hematopoietic, vascular, and perhaps neuroectodermal development (10). Evidence suggests that EWS-FLI1 fusion protein is the key oncogenic lesion in Ewing sarcoma. EWS-FLI1 acts as the dominant oncogene (11). The transcription factor resulting from the translocation replaces the EWS RNA-binding domain by the FLI1-ETS domain with the resulting aberrant transcriptional effects. A number of genes have been identified as direct or indirect regulatory targets of the aberrant protein produced by the EWS-FLI1 translocation (9,12). A major hurdle in the attempts to generate model systems of the Ewing family of tumors is the toxicity of the fusion gene. The identity of the cells from which Ewing tumors originate has remained elusive. The observation of the t(11:22) translocation leading to the EWS-FLI1 expression suggests the existence of a primary cell transformed by EWS-FLI1. Evidence from a genetically defined EWS-FLI1 model system suggests a mesenchymal origin of these tumors (13). Recent publications have shown that the t(11;22) breakpoint location and therefore the exact amino acid composition of the resultant EWS-FLI fusion oncoproteins might have prognostic relevance (14, 15, 16). Therefore, the importance of the availability of specimens for genetic analysis cannot be overemphasized (16).
CLINICAL PRESENTATION
Pain is the most common presenting symptom, noted in approximately 90% of cases. Local swelling or mass effect related to the bone tumor is apparent in a majority of children. A distinct soft tissue mass can be appreciated clinically in one third of cases. Significant limitation of movement has been described in 25% of presentations. Neurologic symptoms or signs occur in 15% of children, either as spinal cord compression or peripheral nerve compression. The latter is most often apparent with lesions of the pelvis or about the knee. Fever is present in 10% of cases and has been related to tumor size and metastatic disease at diagnosis.
The diagnostic evaluation of the patient with Ewing sarcoma is shown in Table 9.1. Laboratory findings may include high leukocyte count, a nonspecific finding indicative of tumor bulk or extensive disease. A high leukocyte count has been related to increased risk of tumor recurrence (16,17). Pretreatment serum lactate dehydrogenase (LDH) is of prognostic significance, and the degree of LDH elevation has been related to tumor volume (17). Positron emission tomography using fluorine-18 fluorodeoxyglucose is being studied for detection of osseous metastases in bone tumors and appears to be a sensitive and specific modality in Ewing sarcoma and PNET.
Table 9.1 Clinical Evaluation of the Patient with Ewing Sarcoma | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
 Figure 9.2 A: Ewing sarcoma of the left radius with (B) extensive infiltration of the surrounding soft tissue. |
The diagnostic features of Ewing sarcoma are radiographically defined as a permeative, destructive lesion of bone. In long bones, the tumor most often presents along the metaphyseal region or within the diaphysis (i.e., at the midshaft level). The periosteum often is displaced by the underlying tumor, resulting in the clinical sign of Codman triangle, representing a bone expansile lesion. Although bone expansion is common, new bone formation beyond the periosteal margin is rare. An associated soft tissue mass is typical, occurring in more than 50% of long bone neoplasms (18,19). Both computed tomography (CT) and magnetic resonance imaging (MRI) appear necessary for most sites and are complementary. Using both studies has added substantially to the determination of disease extent, identifying extraosseous involvement and the degree of marrow infiltration linearly (Fig. 9.2). CT has been valuable in outlining the bone and soft tissue extent of central Ewing sarcoma. Better definition of tumor extent by CT scans is credited with improvement in control of pelvic Ewing sarcoma (20). MRI has added to this definition of tumor (Fig. 9.3) (21). Radionuclide bone scan may also be of value, although it may exaggerate the linear tumor extent. MRI may show edema. Whether direct microscopic extension of tumor is associated with the edema is unknown at present.
 Figure 9.3 A: Ewing sarcoma of the tibia seen on a plain radiograph shows cortical disruption, “onion skinning,” and a suggestion of soft tissue involvement. B,C: Coronal and axial magnetic resonance imaging show extensive soft tissue infiltration. |
Table 9.2 Prognostic Factors in Ewing Sarcoma | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Approximately 53% of Ewing sarcomas have a primary site in an extremity, and 47% have central primaries. Ewing sarcoma presents in the proximal extremities in 20% to 30% and distal extremities in 30% to 40% of cases (22, 23, 24). Metastases are present at diagnosis in approximately 20% to 25% of patients.
Primary lesions of the rib are associated with direct pleural extension and significant extraosseous soft tissue mass in a majority of cases (25,26). The original description of thoracopulmonary malignant tumors by Askin et al. in 1979 characterized small round cell tumors in this location as a separate clinicopathologic entity, often called Askin tumor (27). He described a female predominance and a short median survival (8 months). These tumors tend to have a large soft tissue component that can displace most of one lung with or without much rib involvement. This patient group has special management issues (28,29).
The frequency of overt metastasis is estimated at 25% to 30% for pelvic primaries and less than 10% for tumors of the extremities or ribs. The sites of metastatic disease at diagnosis parallel the distribution noted with treatment failure, most often involving the lungs (40%) or bones (40%), with less common disease involving the bone marrow, lymph nodes, soft tissue, visceral sites, or, rarely, the central nervous system (30). No formal staging system has been recognized for Ewing sarcoma. The disease factors most recognized as prognostically significant include the bone of origin or primary site, older age, tumor size, presence or degree of soft tissue extension, and identification of hematogenous metastasis at diagnosis (Table 9.2). Smaller tumors (less than 200 mL in volume) and distal extremity tumors tend to be favorable (24,31). At present, the most important prognostic factor at diagnosis appears to be the presence of metastasis (32,33). Reports suggest that the response to initial chemotherapy is significant as well (34). The prognostic importance of the EWS-FLI1 fusion in Ewing sarcoma warrants additional study (14,15).
SELECTION OF TREATMENT
Local Control
The relative roles of surgery and radiation therapy for local treatment of Ewing sarcoma have long been debated. In 1953, Wang and Schulz (35) reported 5-year survival in 6 of 36 children treated with wide-field irradiation, as compared to 1 of the 14 treated by primary surgical resection. Using 50 Gy to the entire bone, Phillips and Sheline documented survival in 5 of 21 cases in 1969 (36). After these reports, Ewing sarcoma was generally treated with radiation therapy except for small tumors in expendable bones. Reviews have indicated an overall rate of local tumor control of 75% to almost 95% after primary radiation therapy (3,22,37, 38, 39, 40, 41).
Historically, the use of radiation therapy has been the local treatment of choice. However, the role of surgery has increased because of several developments. These important developments include the recognition that the local failure rate of radiotherapy for Ewing sarcoma ranges from 9% to 25%. In addition, the development of innovative surgical techniques allowing preservation of limb and structural bone function has promoted surgery as an alternative to radiation. The routine use of cytoreductive chemotherapy, as discussed later in this chapter, often produces a significant decrease in the soft tissue component, rendering tumors more readily resectable. In addition, the concern over second malignancies from radiation therapy has prompted the reevaluation of the role of surgery as well (42).
However, the data concerning local control with radiation therapy alone are difficult to interpret. Difficulties in assessing response to radiation therapy are evident in the slow rate of resolution of CT, MRI, and radionuclide bone scan findings after irradiation. The significance of residual soft tissue abnormalities and persistent areas of increased uptake on technetium scans is uncertain (22,23,34,43). In patients who have hematologic metastasis, the implication of residual recurrent tumor at the primary site confirmed by biopsy or autopsy is uncertain because of the possibility of reseeding from the primary tumor (20,44).
Several other fundamental factors make it difficult to interpret the data comparing surgery and radiotherapy for local treatment. The local recurrence rate after radiation is strongly correlated with the primary tumor site. Local failure of extremity lesions is on the order of 5% to 10%, compared with a local failure rate for pelvic lesions of 15% to 70%. Local recurrence after radiation therapy of pelvic lesions is expected to improve with improved tumor imaging and the widespread use of computerized radiotherapy treatment planning. Therefore, old data may be of more limited use in contemporary management. It should be noted that tumor size affects local control. Local control for tumors more than 8 cm in diameter is on the order of 80%, compared with 90% for those less than 8 cm in diameter. The fact that larger tumors are more likely to be treated with radiation instead of surgery influences the clinical results of these modalities.
Some patients are treated with a combination of surgery and radiation therapy. Several factors make it difficult to interpret this data. These include the important effect of quality of radiotherapy on local control. In many “surgical” series, the comparison is not surgery with radiotherapy but rather radiotherapy compared with radiotherapy and surgery. This is analogous to the treatment of many other solid tumors with radiation therapy with resection.
Many surgical series include patients who are at lower risk. They include people who have undergone ray resection of the hands and feet, removal of the wing of the ileum, lower sacrum (lower than the S3 bone), ribs, clavicle, or body of the scapula. One must also consider the relative functional deficits that will be experienced during high-dose irradiation, surgery alone, and surgery with irradiation. In this context, when selecting local treatment for childhood Ewing sarcoma one must consider the rehabilitation capacity and the psychological adjustment of the patient with these local treatments (25,45,46).
Fundamentally, when attempting to compare surgery with radiation therapy in the management of Ewing sarcoma, there is the problem of case selection. Studies are not strictly comparable, so surgery and radiation can be seen not as competitive
but as complementary. However one additional matter must be considered in comparing radiation therapy with surgery: the risk of second malignant neoplasms. As we will describe later in this chapter, radiation and alkylating agents combine to increase the risk of second malignant neoplasms after the treatment of Ewing sarcoma. Studies indicate that the relative risk of sarcomas in the treatment field is related to the radiotherapy dosage and the extent of exposure to alkylating agents.
but as complementary. However one additional matter must be considered in comparing radiation therapy with surgery: the risk of second malignant neoplasms. As we will describe later in this chapter, radiation and alkylating agents combine to increase the risk of second malignant neoplasms after the treatment of Ewing sarcoma. Studies indicate that the relative risk of sarcomas in the treatment field is related to the radiotherapy dosage and the extent of exposure to alkylating agents.
The relative functional deficits that will be experienced during high-dose irradiation, surgery alone, and surgery with irradiation must also be considered. In this context, when one is selecting local treatment for childhood Ewing sarcoma, the patient’s rehabilitation capacity and psychological adjustment should be considered (4,40,47).
Surgery
The role of surgical resection in combined-modality treatment was addressed by the Memorial Sloan-Kettering Cancer Center (23,48). With overall disease-free survival approaching 80% at 3.5 years, Rosen et al. (49) described local treatment failure in 34 (21%) patients after radiation therapy and aggressive multiagent chemotherapy, compared with 0 of 33 patients who underwent surgical resection in addition to chemotherapy. In an influential review of the Mayo Clinic experience with Ewing sarcoma, Wilkins et al. (50) related a significant impact of surgical resection on overall survival: 74% at 5 years, compared with 27% in patients treated without surgery. Other reports have also described improvement in local control with the addition of surgery (22,25,40,45,51, 52, 53). However, the studies reflected selected surgical intervention. For example, in a review by Wilkins et al. (50) of 27 children with microscopically complete resection in a total series of 65 cases, the analysis included 11 patients treated with radiotherapy and no adjuvant chemotherapy. These 11 patients were included in the analysis of the “nonoperated group” and were compared with the patients receiving surgery plus chemotherapy, clearly biasing the results to favor the surgical group. In a more balanced comparison, Brown et al. (22) described improvement in local control with surgery in extremity lesions. There were local recurrences in 0 of 5 resected cases, compared with 3 of 15 patients treated with primary radiation therapy. Brown et al.’s analysis pointed out the selected use of surgery in 35 of 67 patients, clearly selecting cases with distal and smaller lesions for surgery.
Treatment results after surgical resection when combined with chemotherapy have been excellent, even considering the selection factors inherent in surgical subsets. In cases with complete or good partial response to preoperative chemotherapy, Hayes et al. (54) described disease-free survival in 11 of 11 cases with negative operative margins and no added irradiation. Most other series addressing the impact of surgery have incorporated postoperative radiation therapy, often at reduced dosage levels (25,45,47,49,50,53,55). The addition of surgery for “dispensable bones” (e.g., fibula, rib, smaller lesions of the hands or feet) often is considered a significant addition to therapy with little attendant functional deficit, although excellent results have also been reported with irradiation (44). Options for surgical management have also been considered primarily in the very young population, in whom radiation late effects would be more significant (55, 56, 57, 58).
Appropriate comparison of surgical results for Ewing sarcoma entails attention to the adequacy of radiation therapy in the control arm (23,43). The German Cooperative Ewing’s Sarcoma Study (CESS) reported a significant difference in local control and survival favoring the cases operated on in the CESS-81 trial (20,40,55). On review, the high rate of local failure in the radiation therapy group was attributed to a substantial number of cases with inadequate irradiation volume. In fact, a report by Sauer et al. (55), on the same cohort, showed equivalent results of surgery or radiation therapy in small lesions. In addition, the CESS initiated a quality assurance program in 1984. The results of the subsequent study (CESS-86) demonstrate a marked improvement in local control with irradiation compared to the previous trial. From 1986 to 1991, 177 patients with localized Ewing sarcoma were treated with chemotherapy plus radical surgery or surgery plus radiation (45 Gy) or irradiation alone (60 Gy total), with a central treatment planning review for quality assurance. Results are now comparable to those of the surgery group (3-year relapse-free survival 67% after irradiation, 65% after surgery, and 62% after resection plus irradiation) (37,41,43,51,59,60). If the functional results are similar, the potential for second malignancies in patients treated with irradiation for Ewing sarcoma would favor surgery as the treatment of choice for small extremity lesions, dispensable bones, or very young children. High-risk lesions may benefit from combined surgery and radiation (37,61).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


