The pulmonary interstitium is the space between the alveolar walls, which represent the actual functional pulmonary parenchyma. It forms the basic structure of the lung. The basic substance of the interstitium consists of proteoglycans synthesized by fibroblasts and a network of elastic collagen fibers, which account for up to 20% of the dry mass of the lung. Many lung diseases with a wide range of etiologies activate reactions that lead to inflammation of the interstitium with secondary scarring (fibrosis) (Fig. 5.1). As a result, many processes progress to fibrotic healing stages that are then indistinguishable even on histologic examination, not to mention on diagnostic imaging studies (Fig. 5.3). Thus, various disorders eventually lead to pulmonary fibrosis by the common pathophysiologic mechanism of proliferation and activation of fibroblasts. These cells then produce excessive amounts of proteoglycans and collagen fibers. This reaction is physiologically limited; the fibroblasts die by apoptosis as the inflammation heals. However, chronic inflammations or conditions that perpetuate these processes can lead to increasing thickening of the interstitium, resulting in impaired gas exchange. Compliance is also decreased, leading to a restrictive ventilation defect. Advanced cases show fibroblastic invasion of the alveoli with development of fibrotic focal lesions (Fig. 5.2). We first have to answer these questions: Fig. 5.1 The range of etiologies of pulmonary fibrosis. A variety of causes can activate fibrosing processes that then produce similar radiographic findings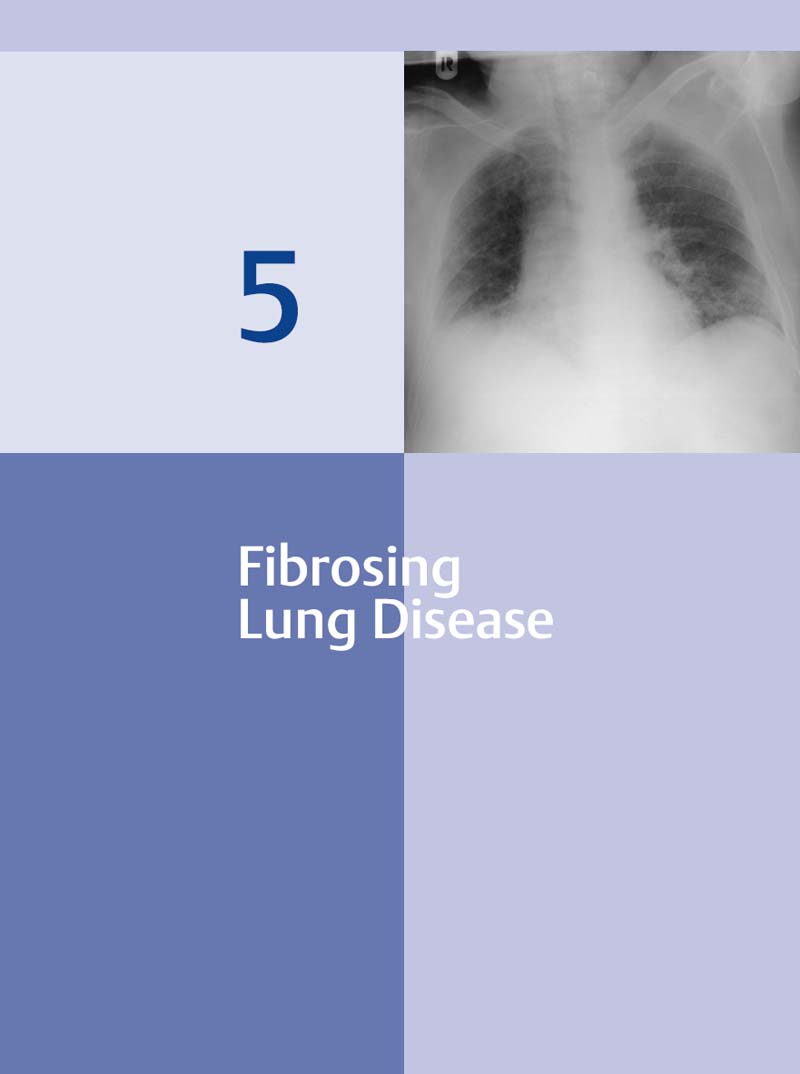
General
 What signs suggest the presence of an interstitial process leading to fibrosis?
What signs suggest the presence of an interstitial process leading to fibrosis?
 More generally: What suggests that interstitial processes are present at all?
More generally: What suggests that interstitial processes are present at all?
 Even more generally: What is the interstitium on the radiograph?
Even more generally: What is the interstitium on the radiograph?
AIP: | Acute interstitial pneumonia |
EAA: | Extrinsic allergic alveolitis |
IPF: | Idiopathic pulmonary fibrosis |
SLE: | Systemic lupus erythematosus |
UIP: | Usual interstitial pneumonia |
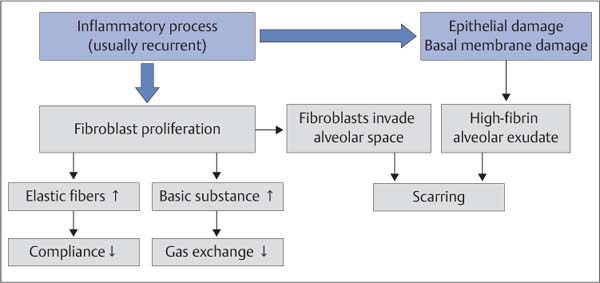
Fig. 5.2 Pathologic mechanism of pulmonary fibrosis.
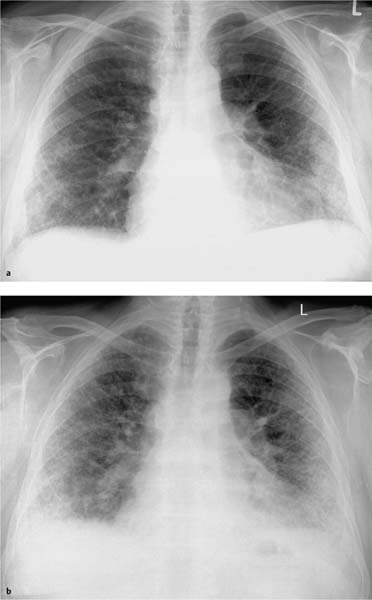
Fig. 5.3a, b Restrictive ventilation defect in idiopathic pulmonary fibrosis.
a Findings include pronounced interstitial shadowing primarily in the mantle of the lung, and a high-riding diaphragm on both sides in broad contact with the heart.
b The follow-up examination 18 months later shows increased shadowing with initial blurring of the diaphragm. Other findings include further thickening of the pulmonary arteries, right heart enlargement, and widening of the upper mediastinum indicative of the onset of cor pulmonale.
Interstitial changes are discrete because of the much smaller volume of intervening tissue (Fig. 5.5) than in the alveolar spaces (Fig. 5.4). In fact, some authors go so far as to question whether it is possible to identify exclusively interstitial processes on chest radiography at all. However, there are in fact phenomena that indicate the presence of such changes. Direct comparison of alveolar and interstitial signs is particularly helpful (Table 5.1). Streaks, lines, and small focal lesions 2–3 mm in diameter (even in a miliary distribution pattern) are regarded as typical interstitial findings (Fig. 5.6). In granulomatosis, these findings exhibit relatively uniform size. They are sharply demarcated and only lose their sharp definition as their size increases (alveolar compression). The proliferation and thickening of the reticular pulmonary shadowing, which can even progress to a coarse weblike pattern, is attributable to a connective tissue reaction. Fine honeycombing is regarded as relatively typical. In contrast to alveolar processes (Fig. 5.7) the interstitial patterns are slow to change. Congestive interstitial pulmonary edema is an exception to this rule.
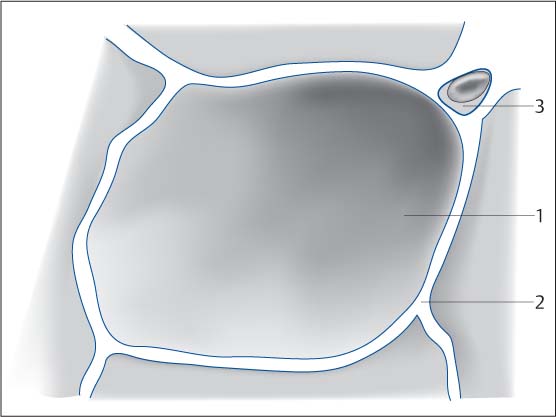
Fig. 5.4 Schematic diagram of the alveolar space. The air-filled space of the alveoli (1) surrounded by the alveolar walls and interstitial septa is far larger than the interstitial space (2). Capillary with erythrocyte (3).
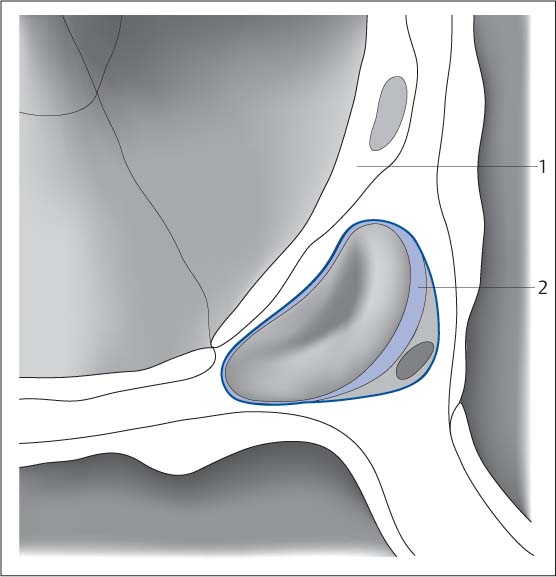
Fig. 5.5 Schematic diagram of the interstitial space. The space between the alveolar walls (pneumocytes [1], functional cells) is referred to as the pulmonary interstitium. It is filled with collagen. Blood vessels (2) and lymph vessels course through this space.
Alveolar | Interstitial |
Acinar foci (5–10 mm) | Small nodules (2–3 mm) |
Ill-defined | Sharply demarcated, uniform |
Gradual transition to normal findings | Streaky and linear densities |
Tendency to become confluent | Sharply demarcated |
Area consolidation | Miliary |
Air bronchogram | Honeycombing |
Alveolar air pattern |
|
Butterfly pattern |
|
Rapid development | Slow development |
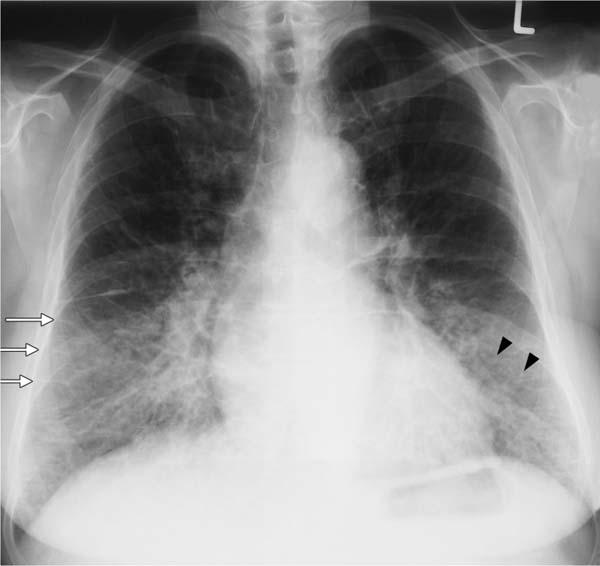
Fig. 5.6 Interstitial pattern in a cardiac pulmonary edema. In addition to pronounced Kerley lines (white arrows) and thickening of the right horizontal fissure due to onset of effusion, findings include acinar nodules (black arrowheads) beginning in the basal segments. Here, only the Kerley lines represent the interstitial pattern. The (discrete) acinar nodules represent onset of alveolar edema.
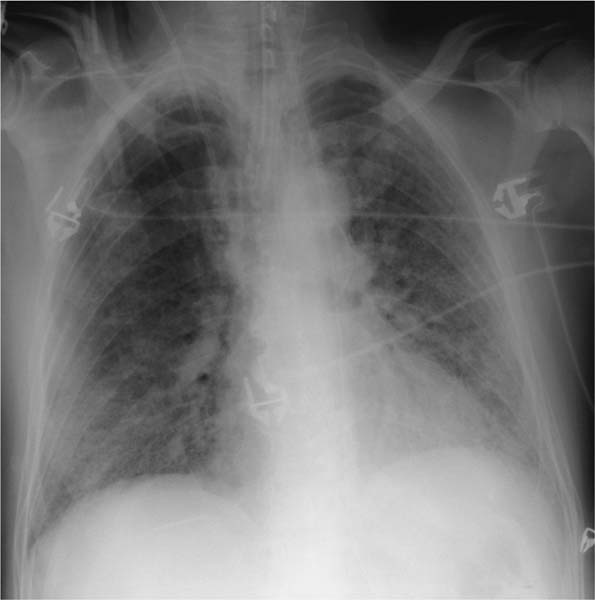
Fig. 5.7 Alveolar shadows in alveolar proteinosis. Alveolar proteinosis may be regarded as a classic example of an alveolar process. The patient is a 56-year-old man who has had a flulike infection and is now presenting with severe respiratory failure. Findings in the intubated patient include bilateral diffuse ground-glass opacities. Heart size is normal. CT demonstrated pathognomonic findings of a leaf pattern, and lavage produced the typical milky exudate.
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is classified as an idiopathic interstitial pneumonia (Table 5.2). It is the most common of this group of disorders of uncertain pathogenesis. Idiopathic pulmonary fibrosis and its acute variant (acute interstitial pneumonia, AIP) also have the worst prognosis.
Radiologic signs of idiopathic pulmonary fibrosis on the chest radiograph include:
 Ground-glass opacities
Ground-glass opacities
 Reticular shadowing
Reticular shadowing
 Disseminated small nodules
Disseminated small nodules
 Progressively reduced depth of inspiration
Progressively reduced depth of inspiration
 Honeycombing
Honeycombing
 Pronounced findings in the mantle of the lung
Pronounced findings in the mantle of the lung
The most important criterion for the differential diagnosis of idiopathic pulmonary fibrosis from secondary fibrosis in chronic emphysematous bronchitis is the progressive reduction in the depth of inspiration (see Fig. 5.3). Decreased compliance leads to a restrictive ventilation defect with a progressively high-riding diaphragm and the picture of a “small lung.” The picture of severe fibrosing bronchitis differs in that it is characterized by a barrel chest deformity. The signs occur in varying severity over the course of the disorder. Diffuse ground-glass opacities of the basal and central segments that blur the vascular structures and pulmonary borders tend to occur early (Fig. 5.8) and in the acute form of the disease. CT detects them in a significantly higher percentage of cases. The onset of fibrosis is marked by fine reticular shadowing in the basal lung segments. This pattern increasingly spreads through the periphery of the lung. Irregular streaky densities and nodular changes occur in the further course of the disorder. The pattern becomes coarsely reticular (Fig. 5.9).
The honeycombing that occurs only in advanced cases is regarded as a typical finding. This pattern is created by a circumscribed cluster of bullae measuring 1 to 10 mm in diameter. These clusters show a predilection for the posterobasal segments (Fig. 5.9b). Note that even advanced cases show neither effusion nor cardiomegaly with signs of congestion such as Kerley lines. However, this picture is often masked by intercurrent infections.
 Notes on Pathology and Clinical Findings
Notes on Pathology and Clinical Findings
Progressive inflammatory processes tending to produce fibrosis of the pulmonary parenchyma (interstitial and alveolar components) that are not the result of extrinsic noxious agents such as pneumoconiosis, extrinsic allergic alveolitis (see below), radiation therapy, etc. or systemic diseases such as scleroderma or systemic lupus erythematosus are classified as idiopathic pulmonary fibrosis (IPF). A further subdivision is still controversial. The Katzenstein classification has recently been expanded by an ATS/ERS study group (Table 5.2). This system differentiates the following forms:
 Acute form (AIP), corresponding to what was once known as Hamman–Rich disease
Acute form (AIP), corresponding to what was once known as Hamman–Rich disease
 Desquamative form with hypercellular alveolar exudate and highly uniform distribution (DIP)
Desquamative form with hypercellular alveolar exudate and highly uniform distribution (DIP)
 A form with a highly irregular distribution of changes and dense focal accumulations of collagen in the interstitium, known as the usual form (UIP or IPF)
A form with a highly irregular distribution of changes and dense focal accumulations of collagen in the interstitium, known as the usual form (UIP or IPF)
 A nonspecific type with diffuse, homogeneous thickening of the alveolar septa: nonspecific interstitial pneumonia (NSIP)
A nonspecific type with diffuse, homogeneous thickening of the alveolar septa: nonspecific interstitial pneumonia (NSIP)
However, various groups (Scadding, Turner–Warwick) have postulated that DIP and UIP may be only different stages of the same disorder, IPF.
Idiopathic pulmonary fibrosis is a disorder typically occurring in advanced age, significantly more often in women than in men. The disorder begins with slight, nonspecific symptoms such as a dry cough that responds poorly to cough medications and dyspnea on exercise. Symptoms progress insidiously over a period of months. Auscultatory findings include inspiratory basal rales. Clubbed fingers occur in up to 50% of all cases. The pulmonary function examination usually shows a purely restrictive ventilation defect. The course of the disease is fatal within 3–5 years in the majority of cases. Only about one-third of patients respond to therapy with steroids or immunosuppressives.
Katzenstein (1998) | ATS/ERS (2002) |
Usual interstitial pneumonia (UIP) | Usual idiopathic pulmonary fibrosis (IPF) |
Acute interstitial pneumonia (AIP) | Acute interstitial pneumonia (AIP) |
Nonspecific interstitial pneumonia (NSIP) | Nonspecific interstitial pneumonia (NSIP) |
Desquamative interstitial pneumonia (DIP) | Desquamative interstitial pneumonia (DIP) |
Respiratory bronchiolitis-interstitial lung disease (RB-ILD) | Respiratory bronchiolitis-interstitial lung disease (RB-ILD) |
Bronchiolitis obliterans with organizing pneumonia (BOOP) | Cryptogenic organizing pneumonia (COP) |
Lymphoproliferative disease (LPD) | Lymphoid interstitial pneumonia (LIP) |
ATS, American Thoracic Society; ERS, European Respiratory Society.
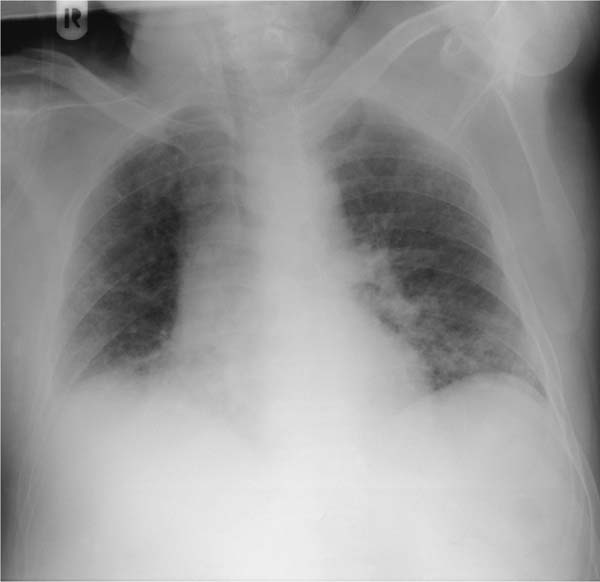
Fig. 5.8 Reduced depth of inspiration in idiopathic pulmonary fibrosis. The patient is a 66-year-old woman presenting with dyspnea on exercise. Diffuse shadowing is seen primarily in the basal segments, which also show hypoventilation and a high-riding diaphragm on both sides. The basal segments exhibit relatively “soft” ground-glass opacities.
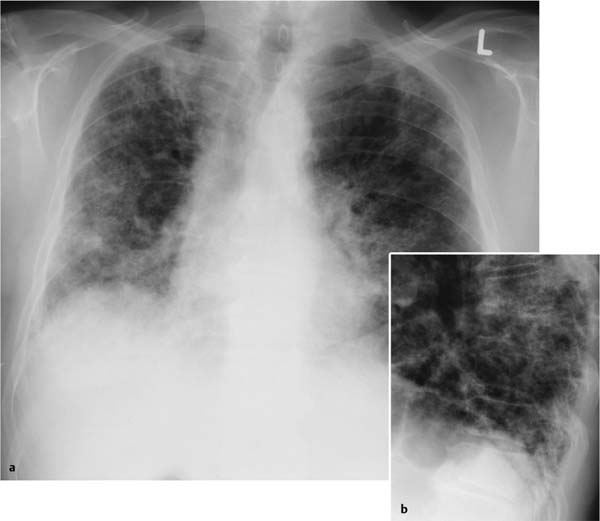
Fig. 5.9a, b Reticular pattern and honeycombing in idiopathic pulmonary fibrosis.
a Increasing pulmonary restriction primarily in the mantle and basal segments of the lung.
b Incipient honeycombing is seen in the posterobasal segments. There are no effusions and hardly any Kerley lines.
On the CT scan, signs of idiopathic pulmonary fibrosis (Fig. 5.10) include increased intralobular and interlobular shadowing with a mantlelike distribution of septal thickening more pronounced in the lower lung fields, isolated nodules, linear densities parallel to the pleura, hyperinflated secondary lobules that can produce an arcade pattern, honeycombing, mosaic perfusion, and secondary changes (pulmonary arterial hypertension, right heart strain, etc.).
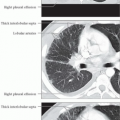
The intralobular shadowing (Fig. 5.11), the occurrence of fine nodular changes, linear densities parallel to the pleura (Fig. 5.12), hyperinflated secondary lobules, occasionally with mosaic perfusion (Fig. 5.13), bronchiectasis adjacent to areas of scarring (Fig. 5.14), and signs of right heart strain are all nonspecific findings. They can also occur in COPD or secondary to pneumonia. However the mantlelike distribution, which primarily involves the basal segments and is particularly well demonstrated on CT, is highly suggestive of idiopathic pulmonary fibrosis. Findings of subpleural arcades (Fig. 5.15) or honeycombing (Fig. 5.16) are essentially diagnostic of the restrictive process.
CT can provide more information about the activity of the fibrotic process than the plain chest radiograph. The fundamental consideration in this context is that the acute process causing the fibrosis (for example, in acute interstitial pneumonia) usually manifests itself as an alveolar pattern with blurring and ground-glass attenuation, whereas the scarring appears as a network of sharply defined markings (Table 5.3).
Lymph node enlargement is inconsistent with idiopathic pulmonary fibrosis. The diagnosis should be reconsidered where such findings occur in the hilum or mediastinum (sarcoidosis? lymphangitis carcinomatosa?). Pleural effusions are other findings that are not attributable to pulmonary fibrosis but can occur in the presence of heart failure.
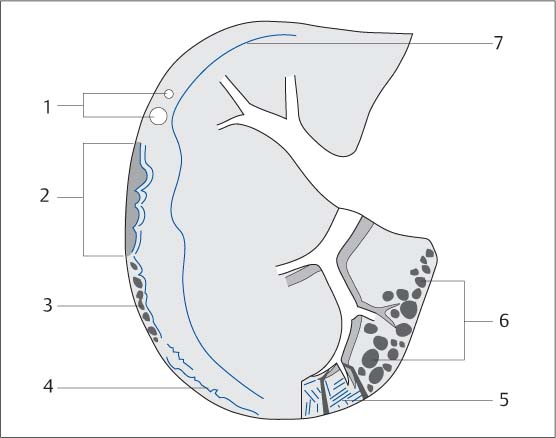
Fig. 5.10 Diagram of the CT signs of a fibrosing pulmonary process.
1 Isolated nodules
2 Arcades
3 Subpleural bullae
4 Linear densities parallel to the pleura
5 Thickened septa and interlobar fissures
6 Honeycombing
7 Peripheral location (mantle of the lung)
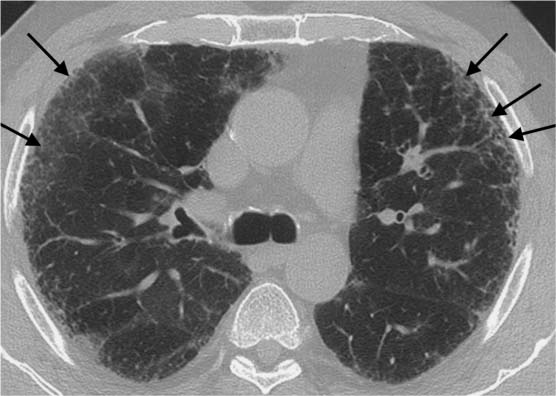
Fig. 5.11 Intralobular shadowing in idiopathic pulmonary fibrosis. The intralobular shadowing (black arrows) shows a mantlelike distribution.
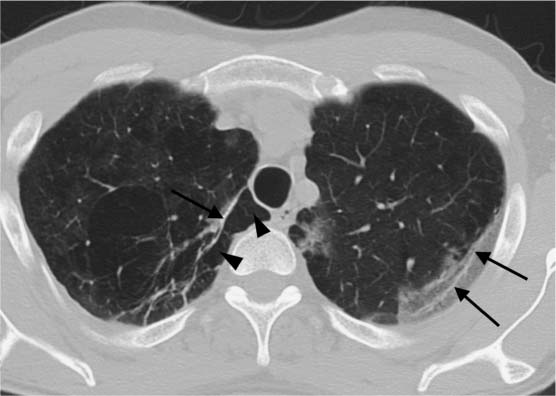
Fig. 5.12 Linear densities parallel to the pleura in idiopathic pulmonary fibrosis. In addition to ground-glass opacities in the left apical mantle, there are linear densities (black arrows) parallel to the pleura with hyperinflation of the subpleural portions of the lung (black arrowheads).
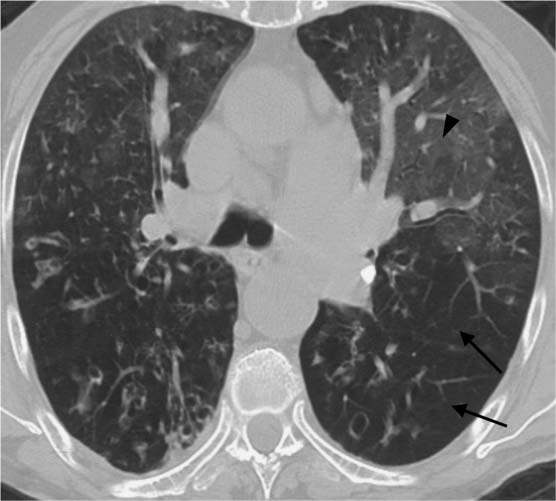
Fig. 5.13 Mosaic perfusion. The patient has severe COPD with advanced emphysema (increased lung volume, peribronchial shadowing, and isolated areas of mucus-filled bronchiectasis). In addition to hyperinflated areas visible as hypodensities (black arrows), findings include hyperperfused areas of higher density showing ground-glass attenuation (black arrowhead).
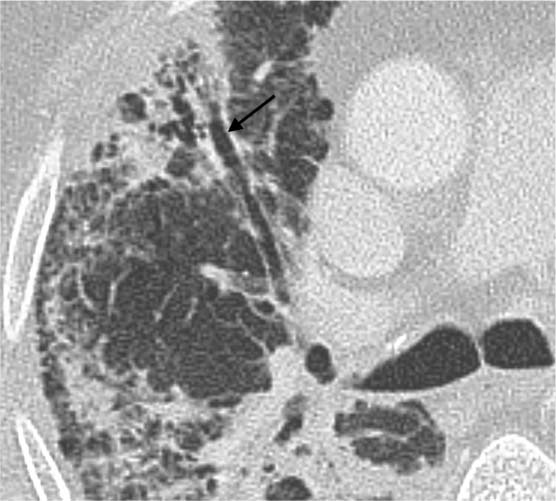
Fig. 5.14 Signs of shrinkage in a fibrotic area. Bronchiectasis (black arrow) and bronchiolectasis indicative of shrinkage of the peripheral area of scarring are non-specific findings. However, in the context of other findings they can be evaluated as a sign of pulmonary fibrosis.
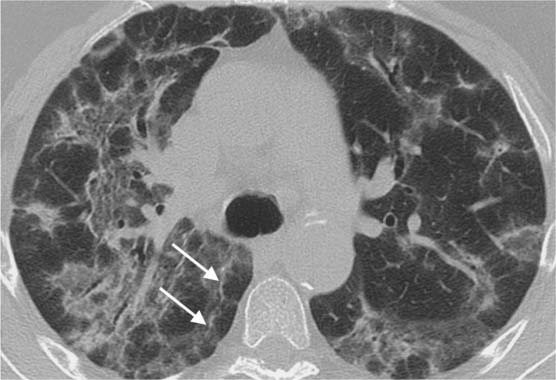
Fig. 5.15 Arcade pattern indicative of pulmonary fibrosis. Findings include significantly hyperinflated secondary lobules, shrinkage of the subpleural pulmonary tissue, and linear densities (white arrows) parallel to the pleura. Here, an arcade pattern is present.
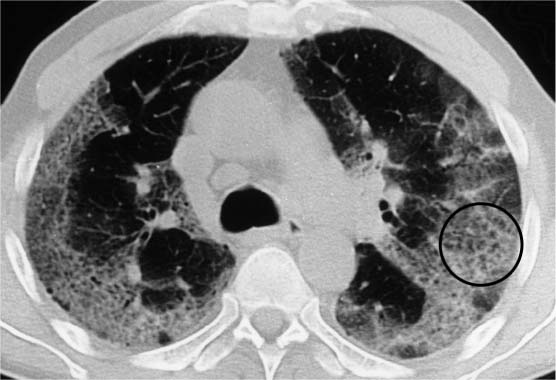
Fig. 5.16 Honeycombing in acute interstitial pneumonia. Extensive consolidations in the mantle of the lung with a significant loss of volume and finely bullous honeycombing (circle).
Acute Interstitial Pneumonia, Hamman–Rich Disease
As early as 1935 Hamman and Rich described a variant of pulmonary fibrosis with a fatal fulminant course in which the fibrotic transformations occurred within the space of a few weeks or months as in a time-lapse film sequence (Fig. 5.17).
Radiographic signs of acute interstitial pneumonia (AIP) include pronounced ground-glass opacities and progressive reduction in the depth of inspiration with severe septal thickening and fine honeycombing typically occurring simultaneously in a rapidly progressive course. In addition to extensive ground-glass opacities in the mantle of the lung (Fig. 5.17 b), Hamman–Rich disease shows septal thickening with hyperinflation of the smallest adjacent alveolar spaces at a very early stage. A fine honeycombing pattern of pulmonary changes rapidly appears even while ground-glass opacities are still present. The ground-glass opacities remain visible as volume loss progresses and architectural distortion increases. CT visualizes the arcadelike pattern of idiopathic pulmonary fibrosis in the mantle of the lung (Fig. 5.17 d).
 In Hamman–Rich disease (acute interstitial pneumonia), ground-glass opacities (signs of activity) can often be demonstrated alongside honeycombing (signs of defects).
In Hamman–Rich disease (acute interstitial pneumonia), ground-glass opacities (signs of activity) can often be demonstrated alongside honeycombing (signs of defects).
Differential Diagnosis of Idiopathic Pulmonary Fibrosis
The most important entity to consider in the differential diagnosis of idiopathic pulmonary fibrosis (idiopathic interstitial pneumonia) is a severe form of chronic emphysematous bronchitis. This differential diagnosis must be made almost daily in clinical practice and is not always a trivial matter. The crucial distinguishing criterion is whether an obstructive pulmonary disorder or a primarily restrictive ventilation defect is present. Signs suggesting obstructive disease include the presence of an emphysematous barrel chest with a high-riding diaphragm, widened intercostal spaces, etc. (see Chapter 2).
Difficult cases to interpret include those where the chronic bronchitis and obstruction are accompanied by factors leading to reduced inspiration, such as ascites, overhydration, or respiratory failure with intercurrent infiltrates. There are also borderline cases in which only the further course of the disorder permits definitive diagnosis. For example, hypoventilation is more rapidly progressive in idiopathic pulmonary fibrosis.
Pulmonary fibrosis in scleroderma closely resembles idiopathic pulmonary fibrosis, although the abnormal findings are rarely as extensive. Cutaneous changes are the crucial distinguishing feature in a differential diagnosis. Esophageal widening is a common finding indicative of systemic involvement.
Fibrosis secondary to tuberculosis is more or less localized and is easier to distinguish. Findings often include a stellate configuration of very dense fibrotic strands. The changes show a predilection for the upper lung fields. They are asymmetrical and can lead to significant destruction of pulmonary tissue with shrinkage. Compensatory emphysema is often observed in such cases. Associated findings include calcification (tuberculomas and hilar lymph node involvement) and calcified indurations of the pleura.
Lymphangitis carcinomatosa is not associated with any destruction of normal pulmonary architecture. The intralobular and interlobular septa are thickened and occasionally show nodular swelling. Pleural effusion indicative of impaired lymph drainage is a common finding here that is rarely seen in idiopathic pulmonary fibrosis.
The fibrotic changes in sarcoidosis, extrinsic allergic alveolitis, and pneumoconiosis are discussed on pages 214, 222, and 234.
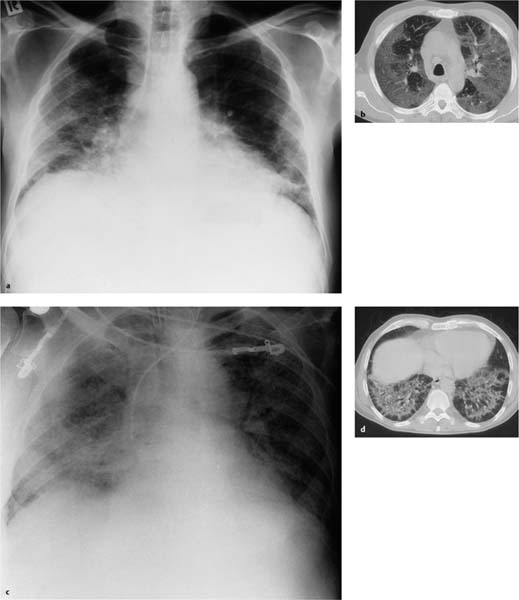
Fig. 5.17 a–d Acute interstitial pneumonia
a Interstitial shadowing is seen in the basal and peripheral areas. Depth of inspiration is significantly reduced, and the heart is in broad contact with the diaphragm. There is no cardiac decompensation.
b The CT scan performed at a relatively early stage of the disorder shows significant ground-glass opacities primarily in the mantle of the lung. Findings are strikingly symmetrical. No pleural effusion.
c, d The further course of the disorder is rapidly progressive. Two weeks later, the patient’s condition has massively deteriorated. Large areas of ground-glass attenuation have developed in both lungs. The patient is intubated. No effusion. Shrinkage in the basal segments has led to detectable areas of subpleural hyperinflation (d) exhibiting an arcade pattern. The patient died a few days later.
Sarcoidosis
Hardly any other intrinsic disorder displays such a variety of symptoms as this generalized occurrence of granulomas of epithelioid cells, giant cells, and lymphocytes. Initially described as a skin disease, the disorder was later named after Boeck.
 The many clinical symptoms and synonyms (sarcoidosis, Besnier–Boeck–Schaumann disease, Boeck disease) are matched by numerous radiographic signs. As a result, the disorder has been called the “chameleon of pulmonary diagnostic medicine” (Fig. 5.18).
The many clinical symptoms and synonyms (sarcoidosis, Besnier–Boeck–Schaumann disease, Boeck disease) are matched by numerous radiographic signs. As a result, the disorder has been called the “chameleon of pulmonary diagnostic medicine” (Fig. 5.18).
Despite the broad palette of radiologic findings, some relatively typical pictures can be identified. Accordingly, the initial diagnosis of pulmonary sarcoidosis is usually made on the basis of incidental radiologic findings, such as on a routine examination prior to employment. In addition to raising initial suspicion, conventional radiography is also an important modality for evaluating the course and activity of sarcoidosis. To a lesser extent, it is also useful in differential diagnosis. This latter application is primarily the domain of CT.
After earlier classification systems, the system of sarcoidosis staging described by Wurm (1958) has gained general acceptance (Fig. 5.19). According to this classification system, three stages are differentiated on the basis of the chest radiograph. Stages II and III are further subdivided. Recently a five-stage classification system (stages 0–4) has come into widespread use (Table 5.4). These stages are also defined on the basis of the plain chest radiograph.
Regardless of the various staging systems, the following section will discuss the relevant radiographic signs in these three groups:
 Radiographic signs of hilar involvement
Radiographic signs of hilar involvement
 Radiographic signs of pulmonary involvement
Radiographic signs of pulmonary involvement
 Radiographic signs of irreversible pulmonary involvement (fibrosis)
Radiographic signs of irreversible pulmonary involvement (fibrosis)
Stage | Findings |
0 | Normal findings |
I | Lymphomas |
II | Lymphomas and parenchymal involvement |
III | Parenchymal involvement only |
IV | Fibrosis |
 Notes on Pathology and Clinical Findings
Notes on Pathology and Clinical Findings
The pathologic substrate of sarcoidosis granulomas consists of nodules of loosely organized epithelioid cells with or without Langerhans cells. Older granulomas hyalinize, although no central caseation occurs as it does in tuberculosis. The etiology of sarcoidosis remains unclear. Formerly, the consensus was that sarcoidosis was closely related to tuberculosis on the basis of the similarity of the pathologic substrates. Sarcoidosis was even thought to represent an abortive benign stage of tuberculosis, a “productive sarcoid form” (manifestations of sarcoidosis often resolve quickly in the presence of florid tuberculosis). However, after the Second World War sarcoidosis came to be regarded as a symptom with multiple etiologies. Today, Uehlinger’s concept of sarcoidosis as a nonspecific hyperergic form of reaction, the morphologic equivalent of an antigen–antibody reaction, has gained general acceptance.
Only syphilis and tuberculosis are comparable to sarcoidosis in terms of the variety of its symptoms with their unpredictable progress, exacerbation, and latency periods. Theoretically, any organ could be affected. Yet aside from skin manifestations, the usually insidious symptoms primarily involve the lymph nodes, eyes, and lungs. Pulmonary involvement in particular often remains clinically silent and is only discovered by chance. The cutaneous sarcoids (lupus pernio, butterfly rash) are regarded as cosmetically disfiguring. Often the differential diagnosis of lymph node swelling that has persisted for years will lead to further diagnostic studies.
In contrast to other generalized afflictions, the complete absence of pain in combination with at most minimal general symptoms (fatigue, loss of appetite, weight loss, subfebrile temperatures) are diagnostic in the presence of extensive examination findings. The relative benignancy of the symptoms in most affected patients contrasts sharply with isolated serious clinical courses involving blindness, severe pulmonary fibrosis, cerebral processes producing the picture of a brain tumor, and the occasional sudden cardiac death.
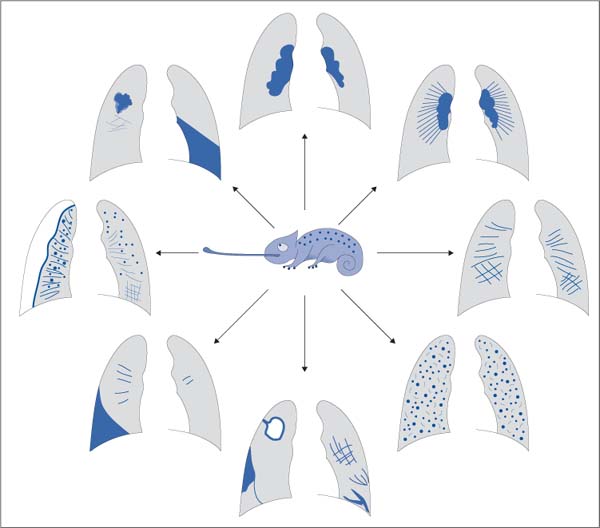
Fig. 5.18 Sarcoidosis, the “chameleon of pulmonary diagnostic medicine.” Hardly any other disorder exhibits such radiographic variation. The broad spectrum of findings includes peripheral and hilar masses, atelectasis, miliary patterns, liquefaction, pleural effusion, pneumothorax, fibrotic pulmonary membrane, and combinations of these findings.
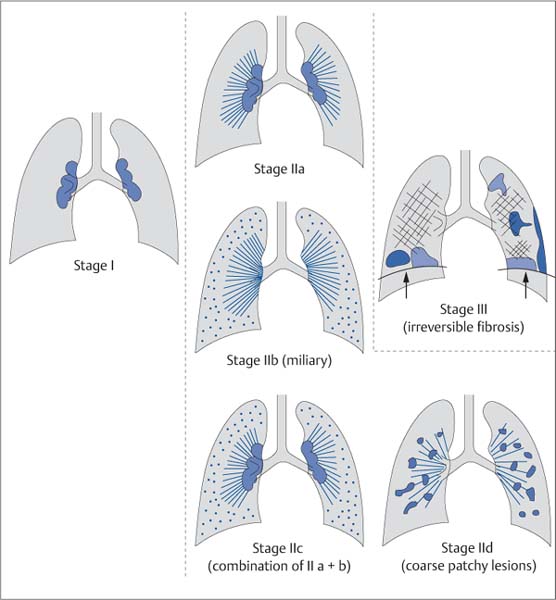
Fig. 5.19 Wurm‘s classification of the radiographic stages of sarcoidosis. The classification system described by Wurm includes three stages. Stage I is characterized by enlargement of isolated hilar lymph nodes; stage III shows signs of irreversible tissue destruction (distraction, shrinkage with reduced depth of inspiration, fibrotic areas, etc.). Stage II is divided into four substages. Stage II a shows a combination of hilar thickening and perihilar streaky densities; stage II b shows perihilar streaky densities with miliary nodules. In contrast, stage II d shows coarse patchy parenchymal involvement.
Hilar Involvement (Stage I)
Hilar involvement in sarcoidosis (approximately corresponding to stage I according to Wurm) shows the following radiologic features:
 Hilar thickening
Hilar thickening
 Usually bilateral and symmetrical findings
Usually bilateral and symmetrical findings
 Radiolucent band along the mediastinum
Radiolucent band along the mediastinum
 Demarcated cardiac border
Demarcated cardiac border
Characteristic findings in stage I according to Wurm include a bilaterally symmetrical pattern of hilar lymph node enlargement. The affected nodes can be massively enlarged. This usually symmetrical bilateral involvement of lymph nodes leads to a widened mediastinum with a polycyclic, sharply demarcated contour.
Differential diagnosis from a lymphoma or bronchial carcinoma (Fig. 5.20) requires attention to the peculiarities of lymph node involvement in sarcoidosis. One relatively characteristic finding is a narrow radiolucent band between the hilum and mediastinum (enlarged lymph nodes in the hilum but not those located in the azygoesophageal recess, Fig. 5.21). A strip of fat is often visible between the lymph nodes and the mediastinal structures. Unilateral hilar lymph node enlargement is rare, occurring in less than 10% of all cases. The lymph node involvement rarely leads to bronchial compression and almost never to atelectasis. This is only observed in endobronchial sarcoidosis. Calcifications are occasionally detectable in the enlarged lymph nodes. These calcifications persist after the swelling of the lymph nodes has receded, creating eggshell calcification figures (Fig. 5.22) that can imitate silicosis. A unilateral pleural effusion is rarely observed in stage I. Such cases usually exhibit a clinical Löfgren syndrome.
The further course of stage I can exhibit:
 Spontaneous healing within 3–12 months
Spontaneous healing within 3–12 months
 Persistence of radiologic findings over several years without any significant changes
Persistence of radiologic findings over several years without any significant changes
 Transition to a chronic form with perihilar fibrosis
Transition to a chronic form with perihilar fibrosis
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


