Splenic Cysts
Non-Neoplastic and Nonparasitic Splenic Cysts
Etiology
Non-neoplastic and nonparasitic splenic cysts are classified into primary (i.e., epithelial, true) and secondary (i.e., pseudocysts, false) cysts, depending on the presence or absence of the internal epithelial lining. Primary or epithelial cysts are considered congenital or developmental in origin. Trauma is the most likely etiologic factor of pseudocysts or secondary cysts, and other causes are considered to be infarction, infection, and pancreatitis.
Prevalence and Epidemiology
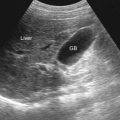
Splenic cysts are uncommon and are usually found incidentally on imaging studies. In one series the incidence at autopsy was 7.6 per 10,000. Epithelial cysts are less common and are mainly seen in children and young adults ( Figures 59-1 and 59-2 ). Pseudocysts constitute 75% to 80% of the splenic cysts ( Figures 59-3 and 59-4 ).




Clinical Presentation
The majority of splenic cysts are asymptomatic, but large cysts may produce nonspecific symptoms such as mild pain, fullness, or discomfort in the left upper quadrant; dyspnea; anorexia; nausea; and vomiting. Although uncommon, acute complications, such as hemorrhage, rupture, or infection, may be the manifesting features. Physical examination may be normal or reveal a left upper quadrant mass with or without tenderness. Results of routine laboratory tests are usually normal.
Pathophysiology
Splenic cysts are usually solitary but can be multiple; 65% are subcapsular, and 80% are unilocular. Cases have been described in accessory spleens as well.
Pathology
Primary splenic cysts have an internal-lining epithelium and can be further subdivided into mesothelial, epidermoid, and dermoid cysts. Mesothelial cysts are considered to be derived from infolding of peritoneal mesothelium or collections of peritoneal mesothelial cells trapped within the splenic sulci during embryogenesis. Epidermoid cysts are lined with squamous epithelium and are thought to develop from metaplasia within mesothelial cysts. The stratified epithelium lining these cysts has immunoreactivity for carcinoembryonic antigen and CA 19-9, and these markers may be elevated in the serum as well. Dermoid cysts are extremely rare, with only a few cases reported, and contain skin adnexa and squamous epithelium. Macroscopically, a glistening inner surface with marked trabeculation is often noted (see Figures 59-1 and 59-2 ). The wall of pseudocysts is composed of dense fibrous tissue, which is often calcified, with no epithelial lining. These cysts contain a mixture of blood and necrotic debris.
Imaging
Although smaller size, internal debris, and calcifications within the fibrous wall are all favorable imaging features that aid in distinguishing false from true cysts, nevertheless both true and false cysts can possess cyst wall calcifications, trabeculations, peripheral septations, and debris (e.g., cholesterol crystals or breakdown products after hemorrhage) ( Tables 59-1 and 59-2 ).
| Modality | Accuracy | Limitations | Pitfalls |
|---|---|---|---|
| Radiography | Poor | Insensitive Nonspecific | Unable to directly visualize splenic abnormality |
| CT | Availability of source literature limited for specifying and comparing accuracy of different imaging modalities for evaluation of splenic abnormality | Radiation exposure Adverse effect of contrast agent | Imaging findings of splenic lesions overlap on noncontrast and postcontrast CT |
| MRI | High cost Patient cooperation | Imaging findings of splenic lesions overlap | |
| Ultrasonography | Operator dependent | Imaging findings of splenic lesions overlap | |
| Nuclear medicine | Poor spatial resolution | Nonspecific | |
| PET/CT | Useful to differentiate, malignant from benign lesion (larger study is needed) | Radiation exposure High cost | False-negative result in splenic metastasis of non–FDG-avid tumors False-positive result in splenic granulomatous disease such as sarcoidosis and tuberculosis |
| Lesion | General Features | CT | MRI | Ultrasonography | Clinical Features | Imaging Features |
|---|---|---|---|---|---|---|
| Cyst | Location: Lower pole, subcapsular Most unilocular Age: True cyst—children or young adults; pseudocyst —<40 yr | Contrast-enhanced: no rim enhancement of the water attenuation, round lesion Rim Ca 2+ : 14% of the true cysts and 50% of pseudocysts | T1 ↓, T2 ↑ | Round, homogeneous, anechoic lesions with marked posterior echo enhancement | No history of underlying malignancy or pancreatitis and/or recent travel to or living in endemic area of Echinococcus A remote history of trauma to the left upper quadrant | Nonspecific cystic lesion |
| Hemangioma | Asymptomatic <2 cm Frequently solitary Age: 35-55 years | Nonenhanced CT: Punctuate or peripheral curvilinear Ca 2+ Contrast-enhanced CT: Variable | T1 ↓↔, T2 ↑ | Well-defined echogenic solid or complex cystic mass | Nonspecific | Nonspecific |
| Hamartoma | Location: Midportion Often solitary | Isoattenuating relative to normal spleen Heterogeneous enhancement | T1 ↓↔, T2 ↔↑ Early phase: Diffusely heterogeneous enhancement Delayed images: More uniform homogeneous enhancement | Solid homogeneous lesion Color Doppler imaging: Hypervascularity | Nonspecific | Relatively similar feature of the normal spleen Delayed phase of dynamic study: More uniform Color Doppler imaging: Hypervascularity |
| Lymphangioma | Location: Subcapsular Age: In children | Single or multiple thin-walled, low-attenuation masses with sharp margins | T1 ↓↑ (bleeding, proteinaceous content), T2 ↑ Enhancing septa | Well-defined anechoic cystic lesions | Nonspecific | Nonspecific cystic lesion |
| Littoral cell angioma | Multiple splenic lesions in patients with hypersplenism Rare | Multiple lesions of similar size, homogeneous enhancement on delayed phase | T1 ↓↔, T2 ↑ Mild heterogeneous enhancement on arterial phase, and homogeneous enhancement on delayed phase | Variable and includes mottled echotexture without discrete lesions | Hypersplenism | Delayed phase: Homogeneously isoattenuating lesion Multiple Splenomegaly |
| Angiosarcoma | Very rare Vigorous prognosis Diffuse involvement common Older patients | Noncontrast CT: Hyperdense area as a result of hemorrhage and Ca 2+ Contrast-enhanced CT: Heterogeneous enhancement | T1, T2: Variable signal intensity, resulting from blood products and necrosis | A complex mass with heterogeneous echotexture | Massive splenomegaly Hemoperitoneum secondary to spontaneous rupture (>30%) | Massive enlarged spleen with heterogeneous enhancement Hemoperitoneum |
| Lymphoma | Most common splenic malignant tumor Splenomegaly not always a reliable sign of splenic involvement | Variable: Splenomegaly without mass, solitary mass, multifocal lesions, or diffuse miliary nodular infiltration | Dynamic study (↓ on 30 sec, ↔ on 2 min), superparamagnetic particles (↑), and diffusion weighted imaging (↑) can improve the evaluation of splenic involvement of lymphoma | Variable | Nonspecific | Nonspecific |
| Metastasis | Metastases uncommon Poor prognosis Solitary or multiple (most common) nodular lesions or diffuse infiltrating lesions | Multiple low-attenuation masses, sometimes solitary | T1↓ , T2↑ | Variable | History of underlying malignancy No evidence of fever | Nonspecific |
Radiography.
Splenic cysts are often large enough to cause splenomegaly and displacement of adjacent organs. Curvilinear or plaque-like calcification is seen on plain radiographs in 5% of true cysts and in 38% of false cysts (see Figure 59-3 ).
Computed Tomography.
Splenic cysts are typically round, well-defined lesions with smooth margins, attenuation near that of water, and a thin or imperceptible wall on computed tomography (CT) (see Figures 59-1 and 59-2 ). Septa may be noted, and rim calcification is seen in 14% of the true cysts and in 50% of false cysts (see Figures 59-3 and 59-4 ). Typically, there is no rim enhancement on a postcontrast scan. Epithelial cysts occurring in an intrapancreatic accessory spleen are frequently misdiagnosed as cystic tumor of the pancreas. In such cases, the presence of thin, compressed splenic parenchyma surrounding the cyst may be noted, suggesting the correct diagnosis (see Figure 59-2 ).
Magnetic Resonance Imaging.
Although splenic cysts typically have signal intensity equal to that of water on both T1- and T2-weighted magnetic resonance imaging, the signal intensity on T1-weighted images may be increased, whereas the signal intensity on T2-weighted images remains high, depending on the composition of the cystic fluid (see Figure 59-2 ). A rim of hypointensity may be caused by calcified wall or hemosiderin deposit at the cyst wall (see Figure 59-3 ).
Ultrasonography.
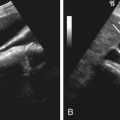
On ultrasonography, typical splenic cysts appear as round, homogeneous, anechoic lesions with marked posterior echo enhancement and with a smooth, thin wall (see Figures 59-1 and 59-2 ). However, sometimes they appear as a more complex picture attributed to thin septations, irregular cyst wall, heterogeneous echo pattern from internal debris or hemorrhage (see Figure 59-4 ), and cyst wall calcifications with bright echo and distal shadowing.
Nuclear Medicine.
Technetium-99m ( 99m Tc)-sulfur colloid scintigraphy shows a defect with thin, rim-like uptake in the periphery.
Differential Diagnosis
The number of diseases that may appear as cystic lesions in the spleen on imaging is extensive. The differentiation between primary and secondary cysts and the differentiation of them from cystic tumors, echinococcal cysts (see later discussion), and abscesses may be difficult radiologically because the findings commonly overlap. The clinical presentation, a history of underlying malignancy or pancreatitis, and or recent traveling to or living in an endemic area of Echinococcus can help narrow the differential diagnoses. A remote history of trauma to the left upper quadrant often can be ascertained.
The evidence of daughter cysts or similar coexisting cystic lesions in other organs such as liver, lungs, brain, and musculoskeletal system are favored for echinococcal cysts. Therefore, imaging survey of these organs may be helpful in suspicious cases. Because cystic metastases to the spleen commonly come from breast and ovarian cancer, followed by melanoma, it may be helpful to survey these organs radiologically to elucidate the possible primary lesions.
Treatment
Medical Treatment.
Secondary cysts, especially those associated with pancreatitis, may resolve spontaneously. However, some will require percutaneous or operative drainage.
Surgical Treatment.
Surgery may be indicated in cases of large symptomatic, mostly primary, cysts. Spleen-conserving surgery may be possible in some cases (e.g., epidermoid cysts arising in accessory spleen).
- •
Cystic splenic lesions encompass various abnormalities, such as neoplasms (including cystic metastases), abscesses, as well as non-neoplastic and nonparasitic cysts.
- •
Clinical findings and history may help narrow the differential diagnosis.
Benign Splenic Tumors
Both benign and malignant primary tumors are rare in the spleen. Among benign tumors, hemangioma is the most common benign primary neoplasm. Other less common benign neoplasms include hamartoma, lymphangioma, littoral cell angioma, hemangioendothelioma, and hemangiopericytoma (see Tables 59-1 and 59-2 ).
Hemangioma
Etiology
Splenic hemangiomas are considered congenital in origin, arising from sinusoidal epithelium.
Prevalence and Epidemiology
Although rare, hemangiomas are the most common benign primary neoplasm of the spleen ( Figures 59-5 to 59-8 ), with a prevalence ranging from 0.3% to 14% in autopsy series. They are found most often in adults 35 to 55 years of age and have no sex predilection. Diffuse hemangiomatosis of the spleen is a rare benign vascular condition occurring as a manifestation of systemic angiomatosis. Associations with Klippel-Trenaunay-Weber, Turner’s, Kasabach-Merritt–like, and Beckwith-Wiedemann syndromes have been reported.




Clinical Presentation
Patients with hemangiomas are generally asymptomatic and have an excellent prognosis. However, large hemangiomas may occasionally manifest as a mass in the left upper quadrant, pain, or splenomegaly. Spontaneous rupture has been reported. Anemia, thrombocytopenia, coagulopathy (Kasabach-Merritt syndrome), and high-output congestive heart failure may rarely occur with large hemangiomas with vast blood flow. Coagulopathy is probably due to sequestration of red blood cells and platelets and to consumption of clotting factors in hemangiomas.
Pathophysiology
Most asymptomatic hemangiomas are smaller than 2 cm, but they can sometimes be huge. Splenic hemangiomas are frequently solitary but may be multiple and also can be diffuse when they manifest as a part of systemic angiomatosis.
Pathology
Histopathologically, splenic hemangioma consists of a nonencapsulated proliferation of vascular channels, lined by a single layer of endothelium and filled with blood. These blood-filled spaces are separated by thin fibrous septa or splenic pulp tissue. The size of vascular spaces in splenic hemangiomas varies, ranging from capillary to, most frequently, cavernous. In diffuse angiomatosis, neoplastic vascular channels may replace the whole spleen. Grossly, splenic hemangiomas will appear as blue-red spongelike nodules in the spleen (see Figure 59-5 ). Smaller hemangiomas, both capillary and cavernous, tend to be solid, whereas large cavernous lesions can develop thrombosis, infarction, fibrosis, and pseudocystic degeneration caused by necrosis. Calcium deposits may be present in the fibrotic area of the mass or in the periphery of the intratumoral cystic spaces.
Imaging
The radiologic appearance of hemangioma ranges from solid to cystic, depending on the gross morphology. Typically, cavernous hemangiomas have a combination of solid and cystic components (see Figure 59-6 ).
Radiography.
On radiographs, large hemangiomas may manifest as a mass in the left upper quadrant or as splenomegaly. When present, multiple small punctuate calcifications or peripheral curvilinear calcifications may be noted.
Computed Tomography.
On unenhanced CT scans, hemangiomas appear as hypoattenuating or isoattenuating, well-marginated masses, sometimes with punctuate or peripheral curvilinear calcifications. Contrast-enhancement patterns can be variable; although strong homogeneous enhancement may be seen immediately after the contrast administration (see Figure 59-7 ), also only low-grade enhancement may be noted compared with strong contrast enhancement of the parent spleen (see Figures 59-5 and 59-6 ). The area of degeneration remains hypoattenuating relative to normal spleen until the delayed phase.
Magnetic Resonance Imaging.
Splenic hemangiomas are mildly low to isointense on T1-weighted images and mildly to moderately hyperintense on T2-weighted images. Contrast-enhancement patterns are similar to those of CT. Compared with hepatic hemangiomas, splenic hemangiomas generally do not demonstrate well-defined peripheral nodules on early postgadolinium-enhanced images (see Figure 59-8 ). Again, this characteristic is thought to reflect the differences in vascular supply to the background organ (spleen vs. liver) rather than inherent differences between splenic and hepatic hemangiomas.
Ultrasonography.
On ultrasonography, a hemangioma may manifest as a well-defined intrasplenic or pedunculated echogenic solid or complex cystic mass (see Figure 59-6 ). However, it also can appear as a low-echoic lesion (see Figure 59-7 ). Echogenic calcifications with acoustic shadowing may be present.
Nuclear Medicine.
Traditionally, it has been noted that a 99m Tc-labeled red blood cell scan demonstrates a perfusion defect and persistent filling on the early and delayed blood pool images, respectively. Nuclear medicine has little role in evaluation of splenic hemangiomas in clinical practice.
Positron Emission Tomography With Computed Tomography.
Most benign splenic tumors such as hemangiomas or hamartomas are expected to be non–fluorodeoxyglucose (FDG)-avid lesions.
Hamartoma
Etiology
Splenic hamartoma is thought to be congenital in origin, reflecting a focal developmental disturbance in the spleen. However, it also has been proposed that splenic hamartoma may be a neoplasm or possibly a posttraumatic lesion.
Prevalence and Epidemiology
Hamartomas are rare benign lesions and are found without age or sex predilection. Review of autopsy series has shown that the prevalence of splenic hamartoma is less than 1%.
Clinical Presentation
Most patients with splenic hamartomas have no symptoms, and these lesions are usually found incidentally at radiologic studies. Larger lesions may manifest as a palpable mass, splenomegaly, or, rarely, rupture.
Hamartomas of the spleen have been associated with hamartomas elsewhere in the body and have been reported in cases of tuberous sclerosis and Wiskott-Aldrich–like syndrome. The association of splenic hamartoma with tuberous sclerosis lends support to the hamartomatous nature of the latter condition. In addition, an association of splenic hamartoma with malignancy has been suggested.
Pathophysiology
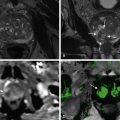
Hamartomas are most likely to occur in the midportion of the spleen, arising from the anterior or posterior aspect of the convex surface ( Figures 59-9 to 59-11 ). They are most often solitary but may manifest as multiple nodules, and lesions up to 19 cm have been reported.

Pathology
Histopathologically, splenic hamartomas are composed of a mixture of unorganized vascular channels lined by endothelial cells and surrounded by fibrotic cords of predominant splenic red pulp with or without (lymphoid) white pulp. Grossly, splenic hamartomas are usually well-circumscribed, solid bulging nodular lesions that tend to compress the adjacent parenchyma. Their gross appearance is typically dark red (see Figure 59-9 ) to grayish white. Despite their well-defined appearance at gross examination, however, hamartomas do not appear well defined at microscopic analysis. Their expansile growth compresses the surrounding red pulp.
Imaging
Radiography.
Splenomegaly may be the only finding of splenic hamartomas on plain radiography, when they are large enough to be visualized.
Computed Tomography.
Although splenic hamartomas may be seen as heterogeneously enhancing lesions on postcontrast CT scans (see Figure 59-9 ), they often can appear as nearly isoattenuating lesions compared with normal spleen before and after enhancement with a contrast agent (see Figure 59-10 ) and, therefore, can be difficult to detect; in such cases, a contour abnormality may be the only finding present. They can also appear as hypoattenuating lesions.
Magnetic Resonance Imaging.
Splenic hamartomas generally exhibit mildly low-signal to iso-signal intensity and moderately high signal intensity on T1- and T2-weighted images, but they may show heterogeneous signal intensity partly because of the varying-sized cystic spaces. If the amount of fibrous tissue is substantial, hamartomas may have regions of low signal intensity on T2-weighted images. The contrast-enhancement pattern is similar to that evident on CT (see Figure 59-11 ).
Ultrasonography.
It has been reported that ultrasonography is more sensitive than CT in detection of splenic hamartomas. The typical appearance is a solid-looking, homogeneous hyperechoic lesion relative to the adjacent normal splenic parenchyma, but some may be seen as isoechoic or low echoic (see Figures 59-9 and 59-10 ). Color Doppler imaging may reveal the hypervascularity of the lesion.
Angiography.
Although the typical hypervascularity of the red pulp within the hamartoma may produce several angiographic findings, such as tumor vessels with aneurysmal dilatation, arteriovenous shunts, vascular lakes, and tumor blush, this examination is seldom performed for diagnostic purposes currently because of the advances in noninvasive imaging methods.
Positron Emission Tomography With Computed Tomography.
Most benign splenic tumors such as hamartomas are expected to be non–FDG-avid lesions (see Figure 59-10 ).
Lymphangioma
Etiology
There is no firm consensus as to the exact origin of splenic lymphangioma, which may represent a hamartomatous rather than a neoplastic lesion. Others have proposed a unified concept of lymphangioma and cystic hygroma as being a congenital developmental defect.
Prevalence and Epidemiology
Splenic lymphangioma is a relatively rare benign tumor with various clinical manifestations that range from an asymptomatic incidental lesion to a large symptomatic mass requiring surgery. Most splenic lymphangiomas occur in children, and adult cases are reported less frequently. Lymphangiomatosis is a syndrome in which multiple organs are involved. Lymphangiomas are either solitary or multiple or may replace the spleen in a condition called lymphangiomatosis. Therefore, in a young patient with splenic lymphangioma, the diagnostic evaluation should be extended to include extrasplenic organs.
Clinical Presentation
In patients with splenic lymphangiomas there is a close relationship between the occurrence of symptoms and splenic size. Larger lesions cause symptoms such as left upper quadrant pain, nausea, and abdominal distention. More extensive or larger lymphangiomas of the spleen may be complicated by bleeding, consumptive coagulopathy, hypersplenism, and portal hypertension.
Pathophysiology
Splenic lymphangiomas cover a broad spectrum, which includes solitary lesions ( Figure 59-12 ), multiple lesions, and diffuse lymphangiomatosis ( Figure 59-13 ). They often involve the capsule and trabeculae of the spleen, where lymphatics are normally concentrated, in contrast to the random localization seen with hemangiomas. Of the solitary subcapsular focal lesions, lymphangioma is the most common, when focal splenic infarction is excluded. With larger multifocal lesions, the tumors are separated by distinct residual splenic tissue. Lymphangiomas can be divided into three types according to the size of the vascular channels: capillary, cavernous, and cystic.


Pathology
At gross examination, splenic lymphangiomas have thick fibrous walls with an internal morphology characterized by fibrous trabeculae (see Figure 59-12 ). Hyalinization and calcification of the fibrous connective tissue may be present. Microscopically, these tumors consist of a single layer of flattened endothelium-lined spaces, filled with eosinophilic proteinaceous material instead of blood as seen in hemangiomas. When the histologic characteristics are not clear, the endothelial origin of the cyst may be established with immunohistochemical techniques that demonstrate reactivity for factor VIII.
Imaging
Splenic lymphangiomas typically show cystlike appearances on cross-sectional imaging studies (see Table 59-2 ).
Radiography.
Splenomegaly may be the only finding of splenic lymphangiomas on plain radiography, when they are large enough.
Computed Tomography.
On CT scans, lymphangiomas appear as single or multiple, thin-walled, low-attenuation masses with sharp margins that are typically subcapsular. No significant contrast enhancement is typically seen (see Figure 59-12 ). Curvilinear peripheral mural calcifications may be noted.
Magnetic Resonance Imaging.
On T1- and T2-weighted MRI, the cystic lesions of variable sizes that correspond to the dilated lymphatic spaces typically have hypointensity and hyperintensity relative to the surrounding parenchyma. However, the T1 signal intensity may be high, resulting from internal bleeding or the presence of large amounts of intracystic proteinaceous content. The intervening septa appear as hypointense bands on both T1- and T2-weighted MRI, corresponding to the presence of fibrous connective tissue. Moderate to intense septal enhancement is seen on a postcontrast study during the late phase.
Ultrasonography.
On ultrasonography, splenic lymphangiomas appear as well-defined anechoic cystic lesions with occasional internal septations and intralocular echogenic debris. Tiny echogenic calcifications may be noted. Color Doppler ultrasonography can demonstrate the vasculature along the cyst walls.
Nuclear Medicine.
Nuclear medicine currently has no role in the evaluation of splenic lymphangiomas.
Positron Emission Tomography With Computed Tomography.
Splenic lymphangiomas are expected to be non–FDG-avid lesions.
Littoral Cell Angioma
Etiology
Littoral cell angioma of the spleen is a rare vascular tumor arising from littoral cells, which line the splenic sinus of the red pulp.
Prevalence and Epidemiology
Littoral cell angiomas may occur at any age and have no gender predilection. Although these tumors were originally thought to be benign, their biologic behavior has not been firmly established because there have been several reports of littoral cell angioma with malignant features. Also, an association between littoral cell angioma and other malignancies, including colorectal, renal, and pancreatic adenocarcinoma and meningioma, has been described.
Clinical Presentation
Typically, patients with littoral cell angioma present with anemia or thrombocytopenia. Other systemic symptoms such as fever, chills, weakness, fatigue, and pain have been reported. At physical examination, splenomegaly is noted. In most patients, splenectomy is performed because symptomatic hematologic problems are present and the imaging findings are nonspecific.
Pathophysiology
Littoral cell angioma varies in size from minute foci to large nodules almost completely replacing the splenic tissue.
Pathology
Littoral cell angioma is a neoplasm with characteristic morphologic and immunophenotypic features distinguished from the other vascular splenic tumors. Microscopically, it is composed of anastomosing vascular channels resembling splenic red pulp sinuses. Immunohistochemically, the neoplastic cells express both endothelial (factor VIII) and histiocytic (KP-1, lysozyme) markers.
Imaging
There is a remarkable uniformity to the clinical characteristics and radiologic findings, and littoral cell angioma usually manifests as multiple splenic lesions in patients with hypersplenism. All modalities including ultrasonography, CT, and MRI usually demonstrate splenomegaly and multiple lesions of similar size and appearance (see Table 59-2 ).
Radiography.
Splenic littoral cell angioma may appear as splenomegaly on plain radiography.
Computed Tomography.
Littoral cell angioma typically manifests as multiple hypoattenuating lesions on CT, but lesions with this appearance have a broad differential diagnosis, including other primary splenic neoplasms, lymphoma, infection, and systemic diseases, such as sarcoidosis. However, on delayed phase contrast-enhanced images, littoral cell angiomas homogeneously enhance and become isoattenuating relative to the remaining splenic parenchyma, a finding that may help limit the differential diagnosis. No capsular calcification or tiny cystic spaces associated with the nodular lesions seen on histologic examination are identified at CT. There is no significant abdominal adenopathy, which is typically seen in patients with splenic metastasis and lymphoma, in patients with littoral cell angioma.
Magnetic Resonance Imaging.
MRI shows the tumor to be multiple with regular well-defined margins and mildly low signal to isointensity on T1-weighted images, moderately high signal intensity on T2-weighted images, mild heterogeneous enhancement on arterial phase imaging, and homogeneous enhancement on delayed phase imaging.
Ultrasonography.
The ultrasonographic pattern of littoral cell angioma is variable.
Positron Emission Tomography With Computed Tomography.
Further studies are warranted to evaluate the role of FDG-PET/CT in the differentiation of littoral cell angiomas of the spleen from other ominous splenic malignancies.
Inflammatory Pseudotumor/Myofibroblastoma
Etiology
The cause of inflammatory pseudotumor is unclear. It has been speculated that this lesion represents a reactive tumor-like condition secondary to the infectious or autoimmune disorder. However, now this lesion generally is considered neoplastic and has been redesignated as myofibroblastic tumor.
Prevalence and Epidemiology
The exact nature of myofibroblastic tumor is unclear. Although it is rare and generally considered benign, it is potentially a locally aggressive lesion. Most patients are adults.
Clinical Presentation
There is either absence of or vague systemic symptoms of fever and malaise with a mass.
Pathophysiology
Myofibroblastic tumors are usually solitary but may be multiple.
Pathology
Grossly, this lesion is well defined, with a great size range from a few centimeters to up to 12 cm ( Figure 59-14 ). Microscopically, the lesion consists of inflammatory (lymphocytes, plasma cells, eosinophils, and histocytes) and spindle cells with varying amounts of granulomatous reaction, fibrosis, and necrosis. The spindle cells have an immunophenotype interpreted as myofibroblastic. The predominant growth pattern may be sclerotic, xanthogranulomatous, or plasma cell granuloma type. Central coagulation necrosis is frequently seen, in association with a neutrophilic infiltration.