Sporadic GIST
Familial GIST
Carney’s triade
Carney-Stratakis-syndrom
NF-1
Median age
̴ 60 years
̴ 40–50 years
<35 years
<25 years
̴ 50 years
Gender predilection
No
No
w > m
No
No
Associated symptoms
No
Hyperpigmentation, urticaria pigmentose, mastocytosis, dysphagia
Paraganglioma, pulmonary chordoma
Paraganglioma
Neurofibroma, skin changes
Mutations
No germ line mutations
KIT/PDGFR
Not known
SDHB/C/D
NF1, Neurofibromin
Inheritance
–
Autosomal dominant
–
Autosomal dominant
Autosomal dominant
Histology
Spindel cell > epithelioid > mixed cell
See sporadic GIST
Epithelioid
See sporadic GIST
Spindle cell
Localisation
Stomach, small intestine, rectum, mesenterial, others
Small intestine, stomach, rarely rectum
Stomach
Stomach
Small intestine
In adult patients, approximately 60 % of GISTs occur in the stomach and 30 % in the small intestine. Other sites of origin are the colon, including the rectum, in approximately 5 % and the oesophagus in approximately 1 % of adult patients. Rarely, GISTs develop outside the gastrointestinal tract in the mesentery, omentum or retroperitoneum. However, most of those extragastrointestinal GISTs are metastatic or may be detached from a gastrointestinal primary source [13, 14].
31.3 Histology
31.3.1 Cellular Origin
Based on their histology, GISTs were originally considered to be derived from smooth muscle. However, they rarely showed clear-cut features of complete muscle differentiation. Additionally, in many cases, their immunophenotypic profile differed from that of leiomyomas arising from other sites (e.g., the uterus or soft tissue ). The understanding of GIST biology changed significantly with the identification of the near-universal expression of the CD117 antigen, also known as proto-oncogene c-kit, in GISTs in the late 1980s [15]. At that time, it was shown that the interstitial cells of Cajal that are part of the autonomic nervous system of the intestine and that serve a pacemaker function in controlling motility express the KIT receptor [16]. Interstitial cells of Cajal have immunophenotypic and ultrastructural features of both smooth muscle and neuronal differentiation. Because GISTs, like interstitial cells of Cajal, express KIT, interstitial cells of Cajal are thought to be the cell of origin. Additionally, as two-thirds of GISTs express CD34, it is postulated that GISTs originate from CD34-positive stem cells within the gut wall differentiating toward the pacemaker cell phenotype with time [17, 18].
31.3.2 Histopathology
The differential diagnosis of a subepithelial tumor arising in the gastrointestinal tract is broad, and histologic findings observed on haematoxylin and eosin-stained sections are not specific for GIST . The cellular morphology of GISTs is mainly divided into three categories, namely the spindle cell type (70 %), epithelioid type (20 %) and mixed type (10 %) [14, 19]. Whereas gastric, small intestinal and colonic GISTs are mostly composed of spindle cell tumours, KIT-negative GISTs are more often of the epithelioid type [20]. The epithelioid variant may show discohesive, hypercellular, sarcomatous morphology with significant atypia and mitotic activity [21] (Fig. 31.1).
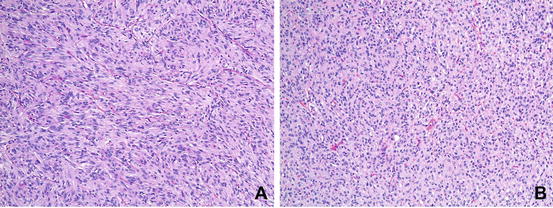
Fig. 31.1
Histologic subtypes of GIST . (a) GIST, spindle cell type. (b) GIST, epitheloid type (Courtesy of Anja Schmitt, MD, Department of Pathology, University Hospital Bern)
31.3.3 Immunohistochemical Features
31.3.3.1 KIT-Positive GIST
A significant breakthrough was the discovery that most GISTs show strong positivity for CD117 (KIT) in contrast to leiomyomas, true leiomyosarcomas and other spindle-cell tumors of the GI tract, which were typically CD117 negative [22]. CD117 is an antigen that is part of the KIT transmembrane receptor tyrosine kinase (RTK) family and is the product of the KIT proto-oncogene (also denoted c-kit). In more than 80 % of GISTs, a mutation in the KIT gene leads to a structural variant of the KIT protein, which is abnormally activated and plays an essential role in cell survival, proliferation and differentiation. When KIT binds to its ligand, it forms a dimer that activates its intrinsic tyrosine kinase activity that, in turn, phosphorylates and activates signal transduction molecules that propagate the signal in the cell (Fig. 31.2).
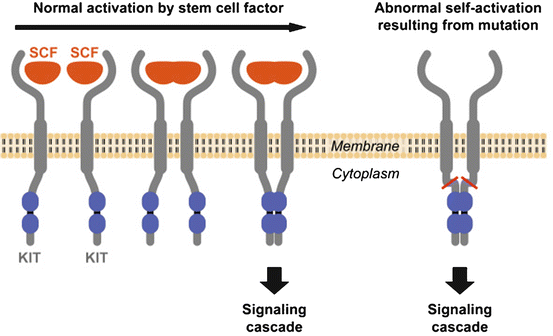
Fig. 31.2
Activation of KIT. Two KIT receptors normally dimerise in the presence of the ligand stem cell factor (SCF) to initiate downstream signalling (left). Mutations in the receptor cause abnormal constitutive signalling without stimulation from the SCF ligand (right) (Hornick JL, MD PhD, Harvard Medical School, Department of Pathology, Boston, MA, and Lazar AJF, MD PhD, Sarcoma Research Center, M. D. Anderson Cancer Center Houston, Texas, reproduced with permission of GIST Support International)
Immunohistochemically, most GISTs (>90 %) show strong positivity for CD117 and usually negativity for desmin and S-100, which are positive in smooth muscle and neural tumours [23]. Although KIT positivity is a major defining feature for GIST , its expression may not be sufficient for diagnosis. KIT-positive malignancies include metastatic melanoma, angiosarcoma, the Ewing’s sarcoma family of tumours, seminoma, and others [24]. Other commonly expressed markers of GIST include CD34 antigen (70 %), smooth muscle actin (SMA; 30–40 %), desmin (<5 %), and S100 protein (~5 %) [25]. In contrast to GIST, leiomyoma and leiomyosarcoma are positive for SMA and desmin and negative for KIT and CD34. Malignant melanoma exhibits diffuse immunoreactivity for S100 protein but can be focally positive for KIT. Schwannomas are strongly and diffusely immunoreactive for S100 protein and negative for KIT [26] (Fig. 31.3).
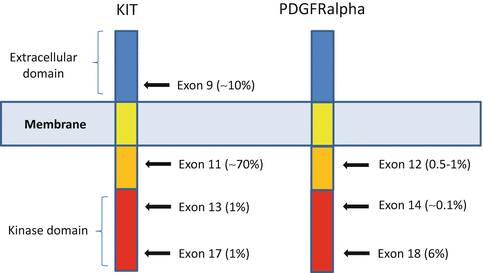
Fig. 31.3
KIT and PDGFRalpha structure (Adapted from Corless et al. Annual Review of Pathology: Mechanisms of Disease 2010)
31.3.3.2 KIT-Negative GISTs
A small subset of GISTs lacks the characteristic KIT mutations [20, 27]. In a proportion of these tumours, activating mutations in the related RTK, PDGFRA, were detected [28]. Many of these PDGFRA-mutant GISTs have an epithelioid morphology. Immunostaining with PDGFRA was shown to be helpful in discriminating between KIT-negative GISTs and other gastrointestinal mesenchymal tumors [29, 30].
DOG1, a calcium-dependent, chloride channel protein, is another highly sensitive and specific marker that often reacts with CD117-negative GISTs [31]. DOG1 expression does not appear to be different between the KIT/PDGFRA mutant or wild-type GISTs. Hence, this marker can be used to diagnose KIT-negative tumour variants.
Inactivation of the succinate dehydrogenase (SDH) complex appears to be an event shared by sporadic and syndromic GISTs that lack mutations in KIT and PDGFRA [32]. Immunohistochemical loss of succinate dehydrogenase subunit B (SDHB) has been shown to be a practical marker to identify SDH-deficient GISTs [33].
The experience with these novel immunomarkers (other than KIT) is currently limited, and problems exist concerning the quality and availability of the commercial antibodies used to stain for them.
31.3.4 Molecular Pathology
Mutational analysis is an essential diagnostic tool in GIST and plays a key role in the confirmation of the diagnosis and in getting prognostic and predictive, hence treatment-relevant—information.
As noted previously, 95 % of adult GISTs overexpress KIT, and approximately one-third of KIT-negative GISTs express DOG1. Therefore, the diagnosis of GIST can be made in most of the cases by observing the macroscopic, microscopic and immunophenotypic characteristics. In cases where the diagnosis of GIST cannot be made based on these features, mutational analysis can be helpful to confirm the diagnosis.
Approximately 80–90 % of GISTs have oncogenic mutations, most of them in KIT and approximately 6–8 % in the PDGFR oncogene. Both of these genes are located on the 4q12 chromosome and encode receptor tyrosine kinases. These oncogenic mutations are the reason for the constitutive activation (“gain of function”) of the respective proteins, leading to uncontrolled stimulation of KIT- and PDGFR-dependent signalling pathways [22].
KIT mutations mostly affect exon 11 and, less commonly, exon 9, 13, or 17 [34] (Fig. 31.4).
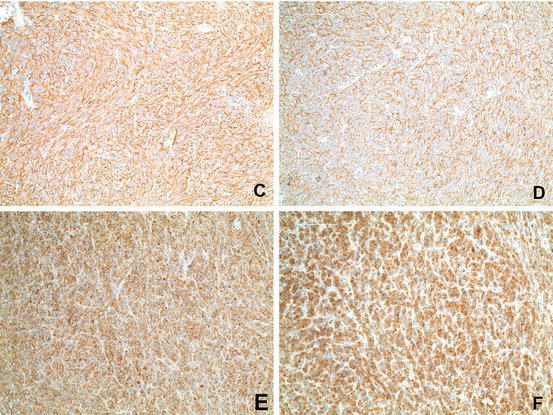
Fig. 31.4
Immunohistochemistry of GIST . (c) Immunohistochemical positivity for c-KIT. (d) Immunohistochemical positivity for DOG-1. (e) Immunohistochemical positivity for CD34. (f) Immunohistochemical positivity for PDGFRalpha (Courtesy of Anja Schmitt, MD, Department of Pathology, University Hospital Bern)
Oncogenic mutations in GISTs include in-frame deletions, missense mutations and tandem duplications. Notably, different mutations are associated with specific tumour locations and maybe clinically more relevant. The prognosis and treatment response correlate with the underlying kinase genotype. Whereas exon 11 mutations are found in virtually every anatomic region, exon 9 mutations are almost exclusively found in intestinal tumours. Tandem duplications are associated with a gastric origin and favourable prognosis. Gastric GISTs with exon 11 deletions have a worse prognosis than those with missense mutations [35, 36]. In terms of the response to systemic therapy, patients with exon 11 mutations are more likely to respond to imatinib than those with other mutations (e.g., in exon 9) or those who lack mutations altogether [37].
PDGFR mutations are mainly located in exons 12, 14, and 18 [38]. A subset of gastric GISTs, particularly tumours with epithelioid morphology, has these types of mutations. The most common mutation is the point mutation D842V, which is relatively insensitive to imatinib although other GIST subtypes confer sensitivity to this agent [28].
GISTs without KIT and PDGFR mutations have been called “wild-type” GISTs, suggesting that these tumours do not have any mutations.
Recently, some GISTs that lack mutations in KIT/PDGFRA have been shown to have inactivation or a deficiency in the SDH complex. Somatic and germline mutations in the genes encoding for the B, C, and D subunits of the SDH enzyme have been described in children and adults with sporadic GISTs that are wild-type for KIT and PDGFRA and those arising in the setting of the inherited Carney-Stratakis syndrome [32, 39].
In a very small population of “wild-type” GISTs, activating oncogenic mutations in BRAF and KRAS have been detected. The clinical relevance of those subentities is unknown, although few data suggest the activity of BRAF inhibitors [40, 41].
Hence, the definition of “wild-type” GIST is changing, and the presence of different new molecular markers has been confirmed. A new definition of “wild-type” GIST was proposed at the ESMO Sarcoma Conference 2014, defining this cohort as lacking KIT exon 9, 11, 13, and 17 and PDGFR exon 12, 14, and 18 mutations.
31.4 Clinical Presentation
GISTs are associated with a broad range of symptoms. Although many smaller GISTs are detected incidentally during endoscopy, surgery or radiologic imaging, others present with various symptoms. Symptoms and signs are not disease specific but are related more to the site of disease. The most common clinical features are the following:
-
Vague abdominal complaints (early satiety, bloating, loss of appetite, nausea, vomiting)
-
Fatigue secondary to anaemia
-
Gastrointestinal bleeding
-
Intraperitoneal haemorrhage
-
Symptoms of obstruction
-
Symptoms of tumour perforation
-
Rarely severe hypoglycaemia due to paraneoplastic tumour production of insulin-like growth factor-2 [42].
Recurrence after primary local treatment is mainly intra-abdominal. The most common site of metastasis is the liver, whereas bone, peripheral skin , soft-tissue and pulmonary metastasis occur much less frequently. Similarly, lymph node metastasis is a very rare condition [43].
31.5 Diagnosis and Staging
The primary investigations before the diagnosis of GIST is made are usually upper or lower endoscopy, abdominal ultrasound or CT. In addition to rectal and liver lesions, where local MRI is much more precise in providing diagnostic and preoperative staging information, the initial modality of choice for staging work-up should include contrast-enhanced abdominal and pelvic CT. The initial work-up should be completed using patient history, routine laboratory testing and chest CT or X-ray [44]. The usual CT appearance of GIST is quite specific and is characterised by a solid, smoothly contoured, soft-tissue mass with heterogeneous enhancement. Larger tumuors may include varying degrees of necrosis and haemorrhage [45].
GISTs are positron emission tomography (PET)-avid tumours. Although routine PET for staging and follow-up is not yet recommended, it could be useful to differentiate an active tumour from necrotic or inactive scar tissue, to reveal a small metastasis that would have been missed otherwise and to determine when early detection of the tumor response to tyrosine kinase therapy is of special concern [46, 47].
Obtaining adequate tumour tissue material for definitive diagnosis before surgical resection has been challenging. Because these tumours tend to be soft and friable, biopsy may cause tumour rupture and may be associated with an increased risk for tumour dissemination. Therefore, preoperative biopsy is not generally recommended if the appearance on CT is highly suspicious of GIST , the tumour is resectable, and the patient is operable. Conversely, biopsy might be needed if radiologic characteristics are atypical, and if preoperative therapy is being considered for unresectable or marginally resectable tumours. As percutaneous biopsy carries the theoretical risk of tumor rupture with peritoneal spread of disease, endoscopic ultrasound-guided biopsy is preferred over a percutaneous one [48, 49].
31.6 Risk Stratification and Stage Classification
Based on three large retrospective trials performed at the Armed Forced Institute of Pathology (AFIP), the tumour size and mitotic rate were identified as the most important prognostic factors [1, 21, 50]. Because this series represents the largest published GIST cohort with long-term follow-up in the preimatinib era, the data formed the foundation for the National Institutes of Health (NIH) consensus approach to risk stratification of GISTs published in 2002 [25].
Subsequently, evaluating long-term follow-up of even more patients, Miettinen et al. suggested new guidelines for the risk stratification, including the primary tumour site as a relevant prognostic factor considering that anatomic location affects the risk for disease recurrence and progression. When using these tools, it is important to appreciate that the mitotic index and tumour size are non-linear continuous variables, so thresholds should be interpreted wisely (Table 31.2).
Table 31.2
AFIP classification
|
Tumour parameter
|
Risk for progressive disease (defined as metastasis or tumour-related death)
|
||||
|---|---|---|---|---|---|
|
Mitotic index (counts per 50HPF)
|
Size (cm)
|
Gastric
|
Duodenum
|
Jejunum or Ileum
|
Rectum
|
|
≤5
|
≤2
|
None (0 %)
|
None (0 %)
|
None (0 %)
|
None (0 %)
|
|
>2 ≤ 5
|
Very low (1.9 %)
|
Low (4.3 %)
|
Low (8.3 %)
|
Low (8.5 %)
|
|
|
>5 ≤ 10
|
Low (3.6 %)
|
Moderate (24 %)
|
n.a.
|
n.a.
|
|
|
>10
|
Moderate (10 %)
|
High (52 %)
|
High (34 %)
|
High (57 %)
|
|
|
≥5
|
≤2
|
None#
|
High#
|
n.a.
|
High (54 %)
|
|
>2 ≤ 5
|
Moderate (16 %)
|
High (73 %)
|
High (50 %)
|
High (52 %)
|
|
|
>5 ≤ 10
|
High (55 %)
|
High (85 %)
|
n.a.
|
n.a.
|
|
|
>10
|
High (86 %)
|
High (90 %)
|
High (86 %)
|
High (71 %)
|
|
According to these guidelines, gastric GISTs that are 2 cm or smaller with a mitotic index of 5 or less per 50 HPF can be regarded as essentially benign, but gastric lesions larger than 2 cm with the same mitotic index have a risk for recurrence. Data are lacking on the prognosis of patients with GISTs smaller than 2 cm with a mitotic count of more than 5 per 50 HPF. Additionally, these data confirmed that small intestinal GISTs are more aggressive than gastric GISTs of equal size This risk classification is an accepted and widely used tool and mainly serves to discriminate patients benefiting from adjuvant systemic therapy [13, 51].
A nomogram was recently published by the Memorial Sloan-Kettering Cancer Center that can be used as an alternative to the risk stratification schema described above. The nomogram can quantify the risk of disease recurrence after complete resection as a continuous variable [52].
Tumour rupture, either at surgery or spontaneously, should be regarded an independent risk factor affecting prognosis negatively [53]. Considering this additional risk factor, Joensuu et al. recently proposed a novel, modified risk classification system by generating prognostic heat and contour maps [54] (Fig. 31.5).
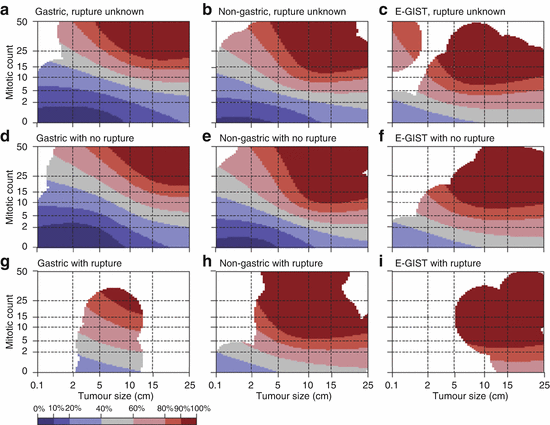
Fig. 31.5
Contour maps for estimating the risk of GIST recurrence after surgery. The upper-row maps are used when the tumor rupture status is unknown (a–c), the middle-row maps are used when the tumor has not ruptured (d–f), and the bottom-row maps are used when tumor rupture has occurred (g–i). Red areas depict high risk, blue areas depict low risk, and white areas indicate a lack of data. The percentages associated with each colour (key) indicate the probability of GIST recurrence within the first 10 years of follow-up after surgery. For example, the middle map of the far left column d shows that the 10-year risk of GIST recurrence of a patient diagnosed with a 10-cm gastric GIST with five mitoses per 50 high power fields (HPFs) of the microscope and no rupture is 20–40 %. The 10-year risk associated with a similar tumour when the mitosis count is ten per 50 HPFs increases to 40–60 %. E-GIST extragastrointestinal stromal tumour (arising outside the gastrointestinal tract) (Reprinted with permission from Lancet Oncology Joensuu et al. [54])
Thus far, mutational status has not been incorporated in any risk classification, although some genotypes have a distinct natural history [44, 55].
Although the TNM classification was published recently, it does not have a clinical impact due to several limitations and, thus, is not recommended [56].
31.7 Management of GIST
For optimal management of GIST patients, it is essential to discuss all relevant information, including medical history and laboratory and radiologic findings, within a multidisciplinary team. Pathologists, radiologists, surgeons, and clinical and medical oncologists should be involved in the decision making to ensure the best treatment strategy for each individual with this disease.
31.7.1 Primary Local Treatment
Complete surgical removal (R0 excision) of localised GISTs is the mainstay of treatment for potentially resectable tumours with a size ≥2 cm [57]. Routine lymph node dissection should not be performed because lymph node metastasis is an extremely rare event [58]. Nevertheless, approximately 50 % of GISTs will recur [43]. Resection can be performed by traditional open surgery or laparoscopic surgery, although the latter approach should only be performed by surgeons with expertise in the laparoscopic management of cancer and mainly for gastric primaries [59]. The importance of achieving negative microscopic margins is a controversially discussed issue because a negative impact on OS in patients treated with adjuvant imatinib is lacking. However, R1 resection may be associated with a greater risk for recurrence [60]. A re-resection in a R1 situation is not mandatory but may be carried out if functional sequelae are not expected. Depending on the primary tumour site (oesophago-gastric junction, small intestine, rectum), neoadjuvant treatment with imatinib should be considered (see Chap. 7.2).
The natural history of small oesophago-gastric and duodenal lesions smaller than 2 cm in size regarding the growth rate and metastatic potential is difficult to anticipate. Many of these lesions will have a very low risk of tumour progression and a low metastatic potential. Endoscopic biopsy may be difficult, and tumour spillage remains a relevant risk. Hence, endoscopic ultrasound assessment and regular follow-up are reasonable in these cases. Should there be any feature of malignant behaviour on ultrasound a resection should also be performed. An algorithmic approach to the management of gastric GISTs based on size and endoscopic ultrasound (EUS) appearance has been proposed [49].
31.7.2 Neoadjuvant Systemic Therapy
The aim of neoadjuvant systemic therapy is to reduce the size of a locally advanced GIST to increase the likelihood of complete resection, reduce surgical morbidity and eventually limit the risk of tumour rupture. Because there are no prospective randomised data, the recommendations on neoadjuvant imatinib therapy are largely based on a few prospective, non-randomised and mainly retrospective studies [61–64].
Eisenberg and colleagues published a prospective phase II RTOG0132/ACRIN6665 trial investigating the feasibility of neoadjuvant imatinib in KIT-positive, resectable ≥5-cm primary GIST , or resectable, recurrent GIST. Sixty-three patients received 600 mg/day of imatinib for 8–12 weeks prior to surgery and then continued imatinib for two additional years. Among the patients with localised primary disease, only 2 (7 %) had an objective response to preoperative imatinib, but stable disease was achieved in 25 (83 %) patients. In 77 % of these patients, complete resection could be performed. The present study confirmed the safety of administering imatinib neoadjuvantly, although the treatment period was quite short [61]. Another open-label, single-arm phase II study from Canada investigated neoadjuvant imatinib treatment with 400–600 mg daily in patients with locally advanced or metastatic GIST that was potentially resectable. Imatinib was administered for a maximum of 12 months to a maximal tumour response. Six of 14 patients showed a partial response, and 8 showed stable disease; no progressive disease was documented. The median treatment duration was 9 months. Therefore, the authors concluded that the optimal preoperative treatment duration should be between 6 and 12 months [64].
Taken all together, the data reveal that there is no consensus regarding the indications for neoadjuvant therapy because a particularly treatment benefit was not proven. However, preoperative therapy is a widely accepted concept, particularly in large, bulky tumours of any origin and notably in GIST arising in the oesophagus, oesophago-gastric junction, duodenum and distal rectum, to reduce significant surgical morbidity. Importantly, a biopsy to confirm the diagnosis and exclude imatinib-resistant mutations is mandatory. The treatment response to imatinib should be evaluated early during the treatment course to exclude tumour progression and prepone resection.
To date, questions regarding the imatinib dose in patients with exon 9 mutation and the duration of additive adjuvant treatment in this specific situation remain unanswered, but a total duration of 3 years appears reasonable.
31.7.3 Adjuvant Systemic Therapy
Although surgery remains the therapeutic modality of choice for localised GIST , the risk of recurrence following complete excision is still eminent. In a recently published analysis of a pool of 2,560 patients, including 10 different population-based published series, the estimated 5-, 10-, and 15-year relapse-free survival [RFS] rates were 71 %, 63 %, and 60 %, respectively [54]. This meaningful risk of recurrence is likely due to persistent microscopic disease following surgery. Therefore, the effect of adjuvant systemic treatment with imatinib has been explored subsequently to improve the likelihood of survival in patients with a high risk of recurrence. However, there is no clear consensus from expert groups regarding the level or cutoff of recurrence risk that would justify the use of adjuvant imatinib [44].
After a few phase II trials with very promising results, the benefit of adjuvant imatinib therapy has been evaluated in at least three randomised studies.
In the multicentre, randomised, double-blind and placebo-controlled US trial Z9001, 713 patients with a resected GIST and a tumour ≥3 cm in size were included and patients were randomly assigned to imatinib 400 mg/day or placebo for 1 year. The study was closed after the first interim analysis, which confirmed a significant reduction in recurrence-free survival that was subsequently the primary endpoint. After a median follow-up of 19.7 months, the 1-year RFS rate was 98 versus 83 % favouring imatinib, with a hazard ratio for RFS of 0.35 and a 95 % CI of 0.22–0.53. A benefit in terms of OS could not be confirmed most likely due to cross-over to active treatment and the short duration of follow-up. Imatinib was well tolerated and showed the known toxicity profile (see below) [65]. That pivotal study led to the accelerated approval of imatinib for the adjuvant treatment of completely resected GISTs ≥3 cm in size. Notably, patients were not stratified according to tumour site and mitotic rate.
The second practise-changing phase III trial was performed by the Scandinavian Sarcoma Group (SSG) XVIII comparing 12 versus 36 months of adjuvant imatinib treatment. Eligible patients were of high risk defined according to the modified consensus criteria as having at least one of the following: a tumour size >10 cm, a mitotic count >10/50 high-power fields (hpf), a tumour size >5 cm with a mitotic rate >5/hpf, or tumour rupture. After recruitment of 400 patients with a median follow-up of 54 months, patients in the 3-year arm showed a significant improvement in RFS, the primary endpoint (5-year RFS, 66 versus 48 %; HR, 0.46; 95 % CI, 0.32–0.65) as well as overall survival (OS, 92 versus 82 %; HR, 0.45; 95 % CI, 0.22–0.89). Subgroup analysis demonstrated that patients with exon 9 or PDGFRA mutation did not show a treatment benefit. In summary, these data established at least 36 months of adjuvant imatinib as a new standard for patients with high-risk GIST [66].
Recently, an abstract of the EORTC 62024 study randomising GIST patients between 2 years of adjuvant imatinib and no adjuvant treatment was presented and showed no significant benefit in the primary endpoint, which was imatinib-free survival, under the intermediate- and high-risk scenario [67]. These results per se implicate that progression of GIST may be delayed but survival might not be improved with the available TKIs.
A few outstanding questions need further investigation. First, whereas there is a consensus that PDGFRA D842V-mutated GISTs should not be treated with adjuvant therapy due to their lack of imatinib-sensitivity, the treatment dose in patients with exon 9 mutation is a matter of debate and 800 mg/day of imatinib may be used analogous to the evidence in the metastatic tumour stage. However, there are often regulatory problems limiting this practise. Additionally, we could not confirm whether “wild-type” GISTs also benefit from adjuvant therapy considering their lower sensitivity to imatinib and more indolent natural history [37, 38, 68].
Second, the question remains concerning the optimal treatment duration and whether treatment should be continued for longer than 3 years. In the Scandinavian trial from Joensuu et al., in both groups, within 6–12 months of discontinuation of adjuvant imatinib, the rates of disease recurrence were similarly increased [66]. Similarly, we know from the BFR-14 trial, in patients with advanced GIST , that some patients who had a complete response to imatinib relapsed even after 5 years of treatment when therapy was interrupted [62]. Hence, the latter findings raises questions as to whether recurrences are truly being prevented or just delayed and whether the duration of adjuvant therapy should be beyond 3 years. Currently, a phase II, non-randomised, open-label multicentre study is investigating 5 years of adjuvant imatinib therapy in patients at significant risk for recurrence following complete resection of primary GISTs (NCT00867113).
Additionally, the optimal treatment duration in the case of tumour rupture is unknown given the uncertainty concerning whether these patients should be viewed as virtually metastatic.
Finally, there is no consensus concerning the definition of high-risk GIST , which depends on different risk classifications.
31.7.4 Systemic Treatment in the Palliative Setting
31.7.4.1 Cytotoxic Chemotherapy
Until 2000, the diagnosis of GIST was not well defined. Therefore, trials published before that time included a mixture of so-called GISTs, leiomyosarcoma and different other sarcoma subtypes, indicating meaningless clinical activity in these patients. Since then, a few trials have investigated the efficacy of cytotoxic chemotherapy in specific GISTs, confirming a very low response rate of 0–5 % [69–71]. As such, overall, the data strongly support the lack of benefit of cytotoxic agents for the treatment of GISTs. Hence, the use of cytotoxic agents is not recommended in daily practise.
31.7.4.2 First-Line Treatment: Imatinib
Imatinib mesylate is a pyrimidine derivative that functions as a specific inhibitor of several tyrosine kinase enzymes, mainly ABL, BCR-ABL, KIT and PDGFR. Imatinib works by binding close to the ATP binding site, locking it and thereby preventing substrate phosphorylation, subsequently leading to the inhibition of signalling pathways involved in proliferation and survival [72, 73].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


