Fig. 5.1
Models of breast cancer metastasis. Serving as a model of metastasis, there are several proposed pathways via which primary breast cancer tumors might metastasize. In the left-most model (1), tumor cells acquire the capacity to metastasize early in the th process of tumorigenesis. Shown in the second model is the tendency for some tumors to produce different clones that each harbor different capacities for metastasis and tissue-specific metastatic proclivities. The next model (3) is a representation of the parellel evolution model. Here, metastatic tumor cells are dispersed from the primary tumor very early and develop separately from and in parallel with the primary tumor. The fourth model depicts the cancer stem cell model in which only stem cells have metastatic capacity (Adapted from Weigelt et al. [13])
As is typically the case with considering a spectrum of diseases as complex as cancer, it is likely that no single model will suffice to explain all of metastasic cancer. What can be said with relative certainty is that metastasis follows a basic set of progressive steps. The basic steps involved in metastasis (Fig. 5.2) are as follows:
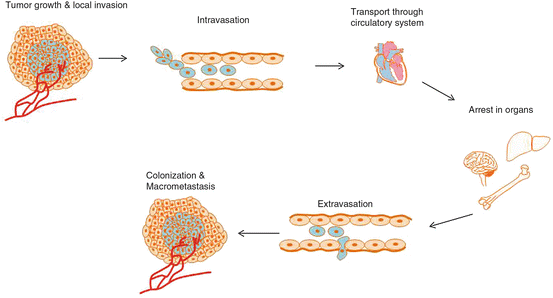
Fig. 5.2
Stages of metastasis. Cancer is generally thought to progress in a step-wise fashion. Tumor cells that acquire the necessary characteristics to “escape” from a primary lesion and locally invade surrounding tissue may then enter into the general circulation via intravasation. From here, tumor cells that survive the harsh environment (shear forces, lack of support structure, growth signals, etc.) can take up residence in distant tissues, again making their way through the endothelial barrier via extravasation. Tumor cells here form micrometastatic colonies that may or may not go on to form macrometsteses
1.
Acquisition of the capacity to invade local tissues
2.
Intravasation (gaining access to the circulation)
3.
Extravasation (exiting from the circulation)
4.
Formation of micrometastasis in a new environment and colonization (growth into macrometastasis)
Each of these steps require the acquisition of a host of specialized characteristics/functions. This chapter will discuss some of the genetic changes that aid cancer cells in their acquisition of these characteristics.
5.3 Stages of the Metastatic Process
5.3.1 Signalling Pathways Involved in Local Invasion
5.3.1.1 Epithelial to Mesenchymal Transition
More than 80 % of cancers are carcinomas; that is they are of epithelial tissue origin. Carcinomas are complex masses of cells, of which as much as 90 % can be non-neoplastic. This diverse collection of non-neoplastic cells compose the tumor stroma. These cells are mostly of mesenchymal origin and are either remnants of the tissue that was invaded by the neoplastic cells or are “recruited” from the surrounding tissue by the neoplastic cells to aid in their growth and survival. Hodgkin’s lymphoma is an extreme example of this phenomenon. In this disease, 99 % of the cells in a tumor are non-neoplastic and surround the rare neoplastic Reed-Sternberg cells.
As is the case in normal epithelial tissue, tumors of epithelial origin rely on heterotypic signalling (signalling between different cell types) between stromal cells and the neoplastic epithelial cells for maintenance of tumor growth and architecture. As the neoplastic epithelial cells proliferate, trophic signals are released and are in turn sensed by cells of the stroma which carry receptors specific for such signals. Thus the tumor and stroma cells proliferate concurrently. These stromal cells can even be found layered within metastases originating from these primary carcinomas, highlighting the interdependence between neoplastic and non-neoplastic cells in a tumor.
The process of epithelial to mesenchymal transition (EMT) involves an alteration in both morphology and gene expression pattern of epithelial cells to that of mesenchymal cells. It is necessary during wound healing to allow re-shaping of the epithelial cell layers and also for some morphogenetic processes of embryogenesis. These are known as type II and type I EMT, respectively [14]. Growing evidence suggests that this process is “hijacked” by cancer cells and used to significantly change their morphology and motility, thereby allowing them to invade nearby tissue. This process is known as type III EMT. It has also been suggested to play a role in cancer progression through maintenance of stem cell-like properties, prevention of apoptosis and senescence, and suppression of immune responses [15]. This is triggered in part by ras oncogene activation within neoplastic tissue cells but also is contributed to by chemical signals from non-neoplastic cells outside the tumor proper.
The leading edges of carcinomas exhibit an EMT front where they are invading surrounding tissue. This can often be seen in immunostained tissue slices containing tumor and non-neoplastic tissue side-by-side. Cancer cells at the edge of the invading tumor do not express epithelial cell surface markers such as E-cadherin, a protein which is strongly expressed by cells in the center of tumors and allows epithelial cells to adhere to one another. Instead, cells express surface markers characteristic of fibroblasts such as vimentin, N-cadherin and fibronectin. Loss of E-cadherin expression through epignetic silencing or expression of mutant forms of this protein has been identified in many carcinoma types and is possibly the single most important change contributing to this type of tumor’s ability to become locally invasive. Several signaling pathways (WNT, TGF-β, FGF, EGF, STAT3 and NF-κB) suppress E-cadherin expression via the transcriptional repressors SNAIL, SLUG and TWIST [7, 16]. The expression of E-cadherin and its associated catenins can also be down-regulated via growth factor mediated-phosphorylation and subsequent proteosomal degradation. These growth factors include epidermal growth factor receptor (EGFR) [17], c-MET (hepatocyte growth factor receptor or HGFR) [18], fibroblast growth factor receptors (FGFRs) [19], Src-family kinases and insulin-like growth factor 1R (IGF-1R) [7]. The degradation of E-cadherin leads to nuclear translocation of β-catenin which affects transcription of genes including the oncogene c-myc and the cell cycle regulator cyclin D1 [16]. The expression of N-cadherin by tumor cells allows them to move into the stroma of the epithelial tissue where other N-cadherin expressing fibroblasts reside. Like E-cadherin, N-cadherin expressing cells bind to one another, however with much less strength than the bonds formed by E-cadherin.
Once these tumor cells escape from the tissue of origin and take up residence in another part of the body, they may find themselves in an environment with a different set of extracellular signals. This may result in a reversion back to the epithelial phenotype, thus becoming more like the cells in the center of the primary tumor from which they originated. This mimics the mesencymal to epithelial transition or MET, which is, like EMT, also involved in wound healing and embryogenesis and may explain why distant metastases often resemble the primary tumors from which they originated. This conversion would also allow cells to regain epithelial cell-cell adhesion and facilitate colonization at new sites [16].
Two other cell transition processes have been described and involve an ameoboid cell phenotype: the collective to ameoboid transition (CAT) and the mesencymal to ameobiod transition (MAT). CAT is caused by β1-integrin inhibition. MAT is triggered by inhibition of proteases and relies on signalling via Rac, Rho/ROCK and EphA2. Ameoboid cancer cells differ significantly from mesenchymal cancer cells. As a result of their unique transition they completely lose cell polarity, are capable of chemotaxis and have very loose attachments to extracellular matrix [16]. They also migrate significantly faster than mesenchymal cancer cells with a speed of up to 20 um/min versus 0.1–1 um/min [20]. They do so by mechanically disrupting matrix structures rather than using proteases to degrade them [21]. Ameoboid cancer cells usually are seen after a patient has been treated with integrin or protease inhibitors. Matrix metalloproteinase (MMP) inhibitors appear to have little to no effect on inhibition of cancer progression in such cases [22, 23].
Transmission of signals between the tissue stroma and tumor is achieved largely via transforming growth factor beta (TGF-β) along with tumor necrosis factor alpha (TNF-α), insulin-like growth factor 1 (IGF-1), epidermal growth factor (EGF) and hepatocyte growth factor (HGF). Interaction between TGF-β and ras oncogenes may trigger EMT. Raf, which is immediately downstream of Ras, can also trigger EMT. Phosphoinositide 3-kinase (PI3K) in turn protects cells from pro-apoptotic functions of TGF-β [1]. TNF-α, produced by inflammatory cells in the early stages of tumor progression, together with TGF-β, are important not only for the initiation but also the maintenance of EMT, via maintenance of NF-κB signalling. NF-κB is a key transcriptional regulator of the inflammatory response and is widely activated in cancer.
In the case of non-epithelial tumors, such as those of hematopoietic and connective tissue and the central nervous system (CNS), the waters are quite muddy. It is possible that an EMT-associated transcription factors are important in the case of CNS, as it is derived from an early embryonic epithelium [24].
Hypoxia and an Activated HIF Program
Hypoxia inducible factor-1 (HIF1) is an oxygen sensitive transcriptional activator and as such is a key regulator for induction of genes that facilitate adaptation and survival of cells from normoxia (~21 % oxygen) to hypoxia (~1 % oxygen). It is composed of two subunits, alpha and beta. The beta subunit is constitutively expressed and the alpha subunit is responsive to oxygen. It is key in the adaptation of cancer cells to hypoxia through its activation of a set of genes that are involved in angiogenesis , iron and glucose metabolism, and cell proliferation/survival (Fig. 5.3). Angiogenesis -associated genes such as vascular endothelial growth factor (VEGF), prostaglandin derived growth factor (PDGF) and angiopoietin-2 are upregulated by HIF-1α. Also upregulated are matrix metalloproteinases 1 and 2 (MMP-1 and MMP-2) and C-X-C chemokine receptor type 4 (CXCR4). While these genes are involved in tumorigenesis, they also serve functions specific to metastasis. MMP-1 helps dissolve the basement membrane and MMP-2 alters architecture of the extracellular matrix. Dissolution of the basement membrane is a key step in migration as it gives tumor cells access to blood and lymphatic vessels in the stroma. CXCR4 in turn causes cancer cells to migrate towards areas of angiogenesis [7]. Inactivation of the p53 signalling system, which would normally activate cell death in conditions of low oxygen, contributes to the ability of cancer cells to survive in a hypoxic environment. Evasion of cell death and the ability to revert to glycolysis for cellular respiration are essential for survival once tumor cells have entered the circulation. Thus characteristics that provide a selective advantage to some cells during tumorigenesis also come in handy once cells exit into the circulation.
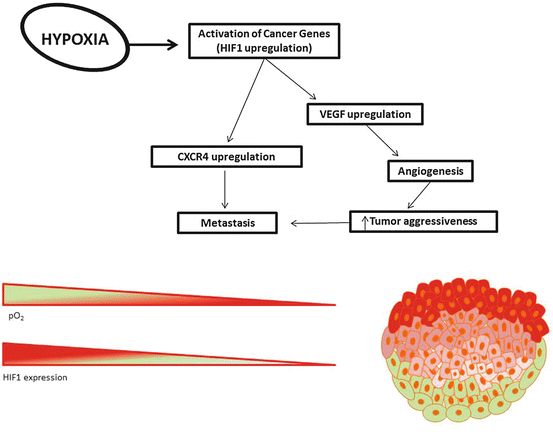
Fig. 5.3
Hypoxia in cancer. Due to rapid proliferation, tumors suffer from a lack of sufficient oxygenation. Cells deeper within the tumor (red and pink cells) have less access to oxygen than those found in the perimeter (green cells). As the partial pressure of oxygen (pO2) drops, HIF1 expression increases. Hypoxia leads to upregulation of many genes involved in metastasis, including CXCR4 and VEGF. CXCR4 expression causes cells to migrate toward areas of angiogenesis and may lead to chemokine-mediated organ-specific metastasis. VEGF upregulation leads to angiogenesis which increases tumor aggressivess as well as the tumor’s capacity for metastasis
5.3.2 Intravasation
The processes of intra- and extravasation are not as well understood as invasion. What is known for certain is that tumor cells encounter unique challenges upon entering the circulation. Most cells require attachment to some kind of substrate for survival and in the absence of such substrate, cells can undergo a form of apoptosis known as anoikis. These circulating cells must also be capable of surviving in the absence of the mitogenic and trophic factors that were present in the stroma from which they originated. Shear forces within vessels can simply tear cells apart. Those that manage to reach larger vessels, some of which may do so by associating with an entourage of platelets, will eventually pass through the heart, after which they will most likely become lodged within the capillaries of the lungs. However, not all metastasis occurs in lungs and thus these cells somehow manage to pass to larger passageways and travel to distant locations in the body. This is likely achieved through arterial-venous shunts. Cells may also pinch off large portions of their cytoplasm and the remaining cell size may be small enough for them to maneuver through the small capillaries. At some point, the cells will need to exit the circulation in some way or another, a process known as extravasation.
5.3.3 Extravasation
In extravasation, we encounter yet another instance of cancer cells hijacking an already existing process for their benefit. Circulating tumor cells express selectin ligands, a group of transmembrane glycoproteins that are also expressed on leukocytes. These proteins are essential for leukocyte transmigration from the circulation to sites of tissue damage or infection, an important component of the body’s adaptive and innate immune response. Selectins expressed on cells that line the vascular walls bind to selectin ligands on leukocytes and cancer cells. This binding is relatively weak and, combined with shear forces in the circulation, results in a sort of rolling movement along the vessels. At some point, a cell or group of cells may become lodged in the vessel. Cells may then proliferate, creating a small tumor that eventually bursts through the vessel wall. Expression of VEGF by cancer cells can also facilitate their extravasation via enhancing endothelial permeability and disrupting the junctions between endothelial cells. Cancer cells with an ameoboid phenotype can easily squeeze through junctions that cells normally would be prevented from traversing. Expression of CXCR4 by cancer cells may result in the selective extravasation of into organs that express CXCL12, such as liver, lung, bone and lymph nodes. Expression of CXCR4 on tumor cells leads to selective extravasation into organs that constitutively express CXCL12 such as liver, lung, bone and lymph nodes [28, 29].
In breast cancer, a gene signature associated with lung metastasis has been identified. Four of the genes in this signature (EREG, MMP1, MMP2 and COX2) have been shown to facilitate blood vessel growth and appear to be essential for extravasation into the lung. Inhibition of these genes resulted in the entrapment of cancer cells within vessels [7, 30]. Again we also see the action of Twist, in this case increasing the ability of cancer cells to migrate intravascularly and extravasate [3, 31, 32].
5.3.4 Colonization and Macrometastasis
After successful extravasation, cells must have the ability to colonize (that is, survive and proliferate) in the new tissue. Antibodies against cytokeratins are used to detect micrometastases in primary carcinoma while epithelial cell adhesion molecule (EpCAM) antibodies can be used to detect micrometastases in lymph nodes. Most extravasated cancer cells do not actually go on to form macrometastases and it can take decades for tumor cells to form clinically detectable metastases after primary tumors are removed [7]. This is referred to as dormancy [33].
The processes involved in this are not well understood. The dormancy period may reflect entry into a state of senescence or may result from active immune surveillance that is able to rid the body of most, but not all, of the cells within micrometastases.
5.4 Evading the Immune System
The body has a number of mechanisms that it uses to ward off cancer development. At the cellular level there is the pRb circuit, DNA repair mechanisms and the apoptotic machinery. At the tissue level, cells that detach from the basement membrane typically undergo anoikis. Until about a decade ago, the role of the immune system in cancer was a highly debated one but evidence of its capacity to identify and destroy cancer cells has been steadily accumulating. First, a body of work in mice provided strong indications for an important role of the immune system in defense against cancer. The development of technology to genetically engineer mice led to the creation of mouse strains deficient in genes that play specific roles in the immune system, such as IFN-γ, perforin, Rag1 and Rag2. These knock-out mice provided key advancements in our understanding of the relationship between the immune system and the development of cancer. But what about humans?
It has been observed that people with compromised immune systems are more likely to develop certain kinds of cancer. Organ transplant recipients, who receive long-term immunosuppressive therapy to prevent rejection of the transplanted tissue, have a very high increased risk of developing some kind of cancer. Cancers of viral origin occur at a much higher frequency in those who are immunocompromised. Kaposi’s sarcoma (caused by human herpes virus 8) occurs in HIV patients at a rate 3,000 times higher than in the general population and tumors caused by human papilloma virus are far more frequent in organ transplant recipients and AIDS patients [24].
The immune system may also be able to recognize tumors of nonviral origin, but it is not clear whether this is indeed the case. Anti-tumor antibodies have also been detected in the blood of cancer patients but it is not known whether these antibodies function in the removal of cancer cells from the body. Another example are tumor-infiltrating lymphocytes which may be recruited to the tumor to aid in its growth or may have invaded the tumor upon recognizing it as “foreign”. The presence of these lymphocytes in several tumor types correlates with improved survival but there is no direct evidence that these are the cause of said improved survival.
The immune system can actively attack circulating tumor cells. For example, natural killer (NK) cells can engage cancer cells via TNF-related molecules such as TRAIL or CD95L, or through the perforin pathway. Both cause tumor cell death, and inhibiting TRAIL or using mice that are deficient in NK cells leads to increased metastasis [7].
5.5 The Role of Cancer Stem Cells in Metastasis
The concept of cancer stem cells (CSCs), first developed over a decade ago, was at first a controversial hypothesis. Accumulated evidence now strongly supports the existence of such cells in a variety of cancers including several leukemias and many solid tumors [34]. The genetic characteristics of CSCs vary by cancer type and even subtype. However, they share in common a high tumorigenic and metastatic potential with unlimited self-renewal capacity. They appear to be resistant to conventional therapies and often able to enter quiescence and/or a state of slow-cycling. This characteristic may explain, at least in part, the dormancy observed in patients whose cancer re-appears decades after initial therapy [33]. It could also explain why CSCs are not as sensitive as other cancer cells to cytotoxic drugs that target actively cycling cells.
This tumor sub-population was named for their similarity to normal adult stem cells present in tissues such as the gastrointestinal mucosa and cells of the hematopoietic system. Due to genetic and epigenetic instability, the CSC population within a single primary tumor is heterogeneous. CSC are not necessarily the “cell of origin” that first gave rise to the primary tumor as cells within the tumor population may undergo changes over time that confer their “stemness”. Another characteristic of CSCs is that they tend to have high expression of EMT markers. Aktas et al. showed that, in patients with metastatic breast cancer, non-responders to treatment had significantly higher expression of EMT markers (62 % vs. 10 % in responders) and ALDH1 (44 % vs. 5 % in responders) [35].
The resistance that CSCs exhibit to conventional drugs may be caused by increased capacity for drug efflux, increased expression of free radical scavengers and increased DNA repair capacity [34]. A great deal of research is now focused on targeting the CSC niche as it appears to be essential for complete eradication of the disease. This has been achieved in part by gene expression profiling of CSCs to identify unique targets. An antibody therapy designed against a CSC-specific isoform of CD44 (CD44v6) resulted in severe skin toxicity in phase I trials for head and neck squamous cell carcinoma (Sauter Riechelmann 2008). Other antibody therapies against markers such as CD123 and CD133 face challenges due to their also being expressed by normal stem cells. Such targets carry a high potential for toxic side-effects, much like traditional chemotherapeutic drugs.
Another method being developed is pre-treatment with a drug aimed at sensitizing the CSCs to conventional therapy. Francipane et al. reported sensitization of colon cancer to chemotherapy after treatment with IL-4 inhibitor [36]. Yet another means of overcoming the resistance of CSCs involves the inhibition of TGF pathway by bone morphogenetic proteins (BMPs). In a mouse xenograft model of brain cancer, this caused differentiation of the CSCs and subsequent cure [37]. Drug efflux pathways may also be targeted to sensitive CSCs to conventional chemotherapy.
5.6 New Targets in the Clinic
As our understanding of cancer has evolved so has the approach to treatment. Although classical chemotherapeutic drugs, radiotherapy and surgical resection are still the most common modes of treatment for most cancer types, there is a trend toward more targeted and individualized therapy. Here we discuss some of the recent developments in treatment specifically targeting metastasis.
Related posts:
 Gastric Cancer: Molecular Mechanisms, Diagnosis, and Treatment
Diagnosis and Management of Gestational Trophoblastic Neoplasia
Neoplastic Epidural Spinal Compression Cord Compression
An Overview of Treatment for Cervical Cancer with Emphasis on Immune Cell-Based Therapies
Gastric Cancer: Molecular Mechanisms, Diagnosis, and Treatment
Diagnosis and Management of Gestational Trophoblastic Neoplasia
Neoplastic Epidural Spinal Compression Cord Compression
An Overview of Treatment for Cervical Cancer with Emphasis on Immune Cell-Based Therapies
 Chemotherapy-Induced Nausea and Vomiting: Molecular Mechanisms and Clinical Approaches
Chemotherapy-Induced Nausea and Vomiting: Molecular Mechanisms and Clinical Approaches
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


