Genitourinary Malignancies
Heiko Schöder
General Considerations
Urologic malignancies account for approximately 25% of all new cancers in the United States (1). The clinical utility of positron emission tomography (PET) with various radiotracers has been studied to some extent for all of these tumors. The diagnostic yield is quite variable for the different malignancies, and the utility of a given radiotracer may depend on the clinical state of the malignancy in question. This chapter discusses some of the most important biochemical alterations in cancer as the basis for PET imaging in urologic tumors. Although most of these data come from experimental work in prostate cancer, much of this likely also applies to the other malignancies discussed in this chapter. In the second part of the chapter the clinical indications for PET imaging and the data of the more important PET studies are discussed in the appropriate clinical context.
Biochemical Alterations as the Basis for PET Imaging in Urologic Malignancies
Fluorodeoxyglucose
Fluorine-18 (18F)-fluorodeoxyglucose (FDG), the agent used for 95% of all clinical PET examinations, traces glucose uptake and phosphorylation by hexokinase. Imaging with FDG takes advantage of the Warburg effect: aerobic glycolysis and an overall increase in glucose metabolism are metabolic characteristics of cancer cells as compared to normal tissue (2,3). The increased glucose metabolism in cancer is mediated through increased expression and activity of glucose transporters (GluT) in the cell surface membrane (4) and through characteristic changes in glycolytic enzyme expression and activity (5,6). Despite the presence of oxygen, glucose is largely metabolized to lactate (aerobic glycolysis). The alterations in glucose metabolism are early events in cancer development (7).
Recent experimental studies have clarified some of the mechanisms and the significance of increased glucose consumption in cancer. In the 1920s, Warburg et al. (2) originally proposed that the increased aerobic glycolysis in cancer cells was the consequence of defects in mitochondrial respiration. Although inhibition of mitochondrial respiration may indeed be one possible explanation (8), more recent theories have invoked alterations in signaling molecules and pathways as important mechanisms for aerobic glycolysis in cancer. One line of evidence has implicated an activation of the hypoxia inducible factor 1 (HIF-1), either as the consequence of intratumoral hypoxia or due to altered gene expression (9). Activation of HIF-1 causes overexpression of numerous proteins, including cell survival and proliferation factors, glucose transporters, and glycolytic enzymes (9). Interestingly, HIF-1 also induces the expression of pyruvate dehydrogenase (PDH) kinase 1, which inactivates PDH, the mitochondrial enzyme that converts pyruvate to acetyl-CoA (10), thereby inhibiting the citrate cycle and oxidative phosphorylation.
Another more recent line of evidence suggests that activation of the oncogene Akt and its gene product, the serine/threonine kinase Akt, may be sufficient to stimulate the switch to aerobic glycolysis (11). Activation of Akt is one of first steps in the PI3K/Akt signaling pathway, which promotes cell growth, survival, and resistance to apoptosis. Activation of Akt kinase in transformed cells stimulates glucose uptake (12) and consumption, and these cells are more susceptible to death than control cells after glucose withdrawal. Activation of the PI3K/Akt pathway causes activation of various other downstream kinases, including mammalian target of rapamycin (mTOR) (7,13). mTOR sensitizes nutrient availability within the cell and regulates cell growth and proliferation (14). In experimental studies, the pharmacologic inhibition of mTOR reverses the cancer-specific increases in glucose metabolism and inhibits the development of prostatic intraepithelial neoplasia (PIN) (7). mTOR may exert some of its effects through activation/stabilization of HIF-1 (15).
Because of the relationship between mTOR and glucose metabolism, PET imaging with FDG is under investigation for determining the treatment response to mTOR inhibitors in various malignancies, including prostate and renal cancers. Unfortunately, this
process is not straightforward because mTOR also regulates Akt through both negative and positive feedback loops (16). This may explain the suboptimal clinical response rates with mTOR inhibitors in some cancers with known activation of the PI3K/Akt signaling pathway. It also highlights the potential limitations of “molecular targeted therapies.” Inhibition of one molecule or pathway may slow down but not necessarily stop the progression of cancer, may only succeed in a subset of patients, or in some cases may even promote cancer progression (17).
process is not straightforward because mTOR also regulates Akt through both negative and positive feedback loops (16). This may explain the suboptimal clinical response rates with mTOR inhibitors in some cancers with known activation of the PI3K/Akt signaling pathway. It also highlights the potential limitations of “molecular targeted therapies.” Inhibition of one molecule or pathway may slow down but not necessarily stop the progression of cancer, may only succeed in a subset of patients, or in some cases may even promote cancer progression (17).
Concerning urologic malignancies, the role of the PI3K/Akt pathway is particularly well documented in prostate cancer. Constitutive activation of this pathway is found in up to 50% of progressive prostate cancers (18) and correlates with a high Gleason score and pathological stage, a higher rate of biochemical recurrence, low response to hormonal manipulations, and progression to metastasis (19,20,21,22). The intensity of Akt expression and the number of Akt expressing cells increases from normal prostate tissue to benign prostatic hyperplasia then to prostate cancer (23).
In addition to Akt, p53 is another frequently mutated gene in cancer. The p53 deficient cancer cells produce similar amounts of adenosine triphosphate (ATP) as normal cells, but significantly higher levels of lactate, a measure of increased glycolysis. The reason is a lack of activation of the gene synthesis of cytochrome oxidase 2 (SCO2), an enzyme complex that is critical during oxidative phosphorylation. This inhibition of oxygen consumption may facilitate the survival of the cancer cell in an hypoxic environment (8). Interestingly, in other model systems, Akt attenuates mitochondrial p53 accumulation (24). This is just one example for many connecting points between various cancer pathways that may affect tumor metabolism and, hence, PET imaging. Indeed, decreased expression of p53 correlates with higher incorporation of FDG into tumor cells (25).
Aerobic glycolysis is not only characteristic for cancer cells, but is in fact mandatory for their continued growth and survival. Although it is possible experimentally to induce a switch from aerobic glycolysis to oxidative phosphorylation in some cancer cells by inhibition of lactate dehydrogenase (LDH), the growth of such cells is clearly inhibited (26). This proves that aerobic glycolysis is not just an epiphenomenon but is indeed necessary for growth and survival of the cancer cell. Among the postulated reasons for increased aerobic glycolysis in cancer is the fact that glycolysis can provide ATP faster that oxidative phosphorylation, and that the products of glycolysis are required for fatty acid synthesis and the maintenance of the nonessential amino acid pool during cell growth (27). In contrast to normal cells, which can switch from glucose consumption to fatty acid oxidation for generation of ATP depending on the substrate availability (28), Akt-expressing transformed cells are limited in their ability to do so (12). One of several possible mechanisms for suppressed β oxidation in cancer could be the Akt-dependent inhibition of CPT-1, the shuttle needed for transporting free fatty acids into the mitochondrion (12).
Before aerobic glycolysis can even start, transport across the cell membrane is the first rate-limiting step for sugar metabolism in cancer cells. Almost 20 years ago, experimental studies in fibroblasts showed that malignant transformation by src and ras oncogenes causes a rapid increase in glucose uptake and GluT-1 expression (29). This is a critical step in the survival of the cancer cell. Whereas normal mammalian cells depend on extracellular signals for maintaining cell surface transporters, growth, proliferation, and survival, cancer cells function autonomously. Despite a lack of external growth factors and survival stimuli, the constitutive expression of Akt in cancer cells can prevent apoptosis, in part by maintaining transporter molecules in the cell surface, such as GluT-1 and amino acid transporters.
Overexpression of glucose transporters (e.g., GluT-1, GluT-3, and GluT-12) has been documented in many malignancies (reviewed in Macheda et al. [4]), including urologic cancers. In prostate cancer, GluT-1 and GluT-12 are overexpressed (30,31), and GluT-1 is expressed to a higher degree in poorly differentiated prostate cancer cell lines, such as DU145 and PC3, than in hormone-responsive LNCaP cell line (31). In clinical prostate cancer specimens, the level of GluT-1 expression increases with advancing grade of malignancy (31).
Overexpression of GluT-1 also occurs in approximately 60% of bladder cancers, but not normal bladder tissue or benign papillomas (32). Among bladder cancers, invasive transitional cell carcinomas and tumors with a high nuclear grade exhibit a higher grade of Glut-1 protein expression than superficial bladder cancers and tumors with lower nuclear grade (32). In bladder cancer, the number of cancer cells with high GluT-1 expression shows an inverse relation with disease-specific and overall survival (33,34).
Increased glucose metabolism in cancer can also be a consequence of hypoxia (anaerobic glycolysis), which is a common feature in many malignancies and leads to overexpression of GluT proteins or alteration of GluT functional activity (34,35,36) and also causes overexpression of hexokinase II (37). Hypoxia is common in bladder cancer (34,38) and prostate cancer (39) and is a marker of poor prognosis. In bladder cancer, the severity of hypoxia correlates with the expression of GluT-1 (34). Of note, while hypoxia is associated with resistance to both radiation and chemotherapy, the inhibition of GluT in hypoxic cells can resensitize those cells to chemotherapeutic drugs and initiate their apoptosis (40), again highlighting the significance of glucose metabolism for the survival of the cancer cell.
For clinical PET imaging with FDG in urologic cancers, some organ-specific mechanisms need to be considered. Prostate cancer differs from other urologic malignancies because it is a hormone-dependent tumor. In the early stages of prostate cancer, FDG uptake may be regulated by the level of androgen stimulation and can be suppressed by androgen withdrawal (41,42). In logical extension of these experimental data, clinical observations suggest that the appearance of lesions with abnormal FDG uptake in patients on antiandrogen therapy indicates a transition to the castrate-resistant state (Fig. 8.18.1) and a worsening of the patient’s prognosis. Moreover, in prostate cancer models, rapidly dividing DU145 prostate cancer cells depended on high levels of glucose, whereas relatively slow-growing LNCaP cells are much less dependent on glucose (43). These data may explain why prostate tumors with high Gleason scores are more likely to exhibit adequate FDG uptake than those with lower scores. They also explain why FDG may not be a good tracer for the diagnosis of primary prostate cancer but is a suitable agent for disease detection and treatment monitoring in patients with progressive castration-resistant metastatic disease.
Renal cancer shows a peculiar molecular abnormality that may have consequences for PET imaging with FDG. About 60% of sporadic renal cell carcinomas are clear cell carcinomas, showing a loss of the von Hippel-Lindau (VHL) tumor suppressor gene (44).
Under normal conditions, VHL binds to the transcription factor HIF-1 and promotes its degradation. Loss of VHL causes HIF-1 accumulation (due to lack of breakdown), which then leads to overexpression of numerous proteins, including growth factors, glucose transporters, and glycolytic enzymes (9).
Under normal conditions, VHL binds to the transcription factor HIF-1 and promotes its degradation. Loss of VHL causes HIF-1 accumulation (due to lack of breakdown), which then leads to overexpression of numerous proteins, including growth factors, glucose transporters, and glycolytic enzymes (9).
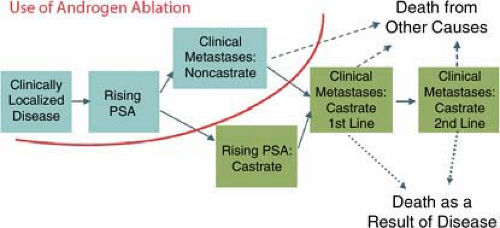 Figure 8.18.1. Model of prostate cancer states. Progressive prostate cancer can be characterized according to well-defined clinical states, which may require different therapies. Use of proper terminology is necessary when comparing the results of clinical and imaging studies in progressive prostate cancer. PSA, prostate-specific antigen. (Figure courtesy of Howard Scher, MD, at MSKCC. Reprinted with permission.) |
In experimental studies, HIF has been established as the key factor in promoting malignant transformation of kidney cells (45). HIF-dependent activation of glycolysis may be one reason for increased FDG uptake in a subset of renal cancers, which in fact occurs in a similar fraction (about 60%) of cases. HIF-1 expression in VHL deficient renal cancers is promoted by mTOR, one of the aforementioned signaling kinases (18). Consequently, the treatment with mTOR inhibitors suppresses the expression of HIF and the production of protein products of HIF target genes, such as GluT and glycolytic enzymes, which can be shown in cell cultures and imaging studies. Treatment with mTOR inhibitors (but not other chemotherapeutic drugs) causes a rapid and significant decrease in FDG uptake in VHL-deficient tumor xenografts. These preliminary experimental data provide some rationale for the use of FDG to identify VHL-deficient (i.e., HIF-dependent) renal cancers and monitor the response to novel anticancer drugs.
In summary, increased glucose uptake and metabolism, as well as some of the underlying molecular mechanism, are well documented in urologic malignancies. This highlights the significance of cancer imaging with FDG PET. Unfortunately, for clinical PET imaging of urologic malignancies, FDG has some limitations related to its physiologic excretion through the urinary tract. Moreover, FDG is not a suitable radiotracer for slow growing malignancies with low glucose metabolism (e.g., early prostate cancer) because the signal will be low and not detectable against background activity. In contrast, this tracer can provide clinically meaningful information in patients with more aggressive disease. This also includes a subset of patients with prostate-specific antigen (PSA) relapse and shorter doubling times, individuals with castrate-resistant metastatic prostate cancer, and patients with recurrent or metastatic renal, bladder, and testicular cancers.
In the interpretation of imaging studies, it should be recognized that aerobic glycolysis is a phenomenon of the cancer cell per se and may not involve the surrounding stroma that continues to rely on oxidative phosphorylation for the supply of energy (46). Accordingly, lactate released as the end product of aerobic glycolysis from cancer cells may be metabolized further by the surrounding stroma, enter the microcirculation (especially when local concentrations are high), or may cause normal cell death by collapse of the transmembrane proton gradient and a breakdown of the extracellular matrix (47). Because current PET scanners cannot distinguish between cancer and stromal cells, the signal obtained always reflects an admixture of metabolic events within a given tumor.
The image quality of FDG PET studies has markedly improved over the past 10 years so that some negative reports in the literature may be outdated. In particular, the routine use of iterative reconstruction algorithms and combined PET with computed tomography (CT) has led to a marked improvement in interpretation of PET studies in the abdomen and pelvis. Nevertheless, the unavoidable urinary excretion of FDG may sometimes interfere in the assessment of prostate, renal, or bladder cancer.
Acetate
Carbon-11 (11C)-acetate is an established agent for tumor imaging. Good tumor-to-background ratios and little urinary excretion make it a suitable agent for imaging urologic malignancies. Acetate can also be labeled with 18F, but methods for safe and efficient synthesis are still under investigation.
The biochemical rationale for cancer imaging with [11C]-acetate is found in characteristic alterations in fatty acid metabolism, which are best documented in prostate cancer but may also apply in other malignancies. Under normal circumstances, exogenous acetate is activated to acetyl-CoA, which can enter various pathways, but predominantly the citrate cycle. In the prostate, normal glandular tissue in the central zone oxidizes citrate like many other mammalian tissues. In contrast, in the epithelial cells of the peripheral zone, the enzyme m-aconitase, responsible for conversion of citrate to isocitrate, is inhibited by high levels of zinc (48).
Testosterone stimulates the key enzyme pyruvate dehydrogenase in prostate tissue, which converts pyruvate to acetyl-CoA, the building block for citrate (49). High concentrations of citrate thus accumulate in the normal prostate and are thought to be essential for maintaining the pH and serve as an energy source for seminal fluid. This has been established in various in vitro studies as well as clinical studies using magnetic resonance (MR) spectroscopy (50,51).
In prostate cancer, inhibition of m-aconitase is no longer present and citrate levels decline. Conceivably, this would suggest increased acetate metabolism through the citrate cycle. However,
in vitro studies tracing the metabolic fate of [14C]-acetate in tumor cell lines (52) revealed that tumor cells incorporate 14C activity mostly into phosphatidylcholine and neutral lipids, and this incorporation showed some correlation with growth activity. Only a small fraction of 14C-acetate was converted into carbon dioxide and amino acids. This observation is in keeping with earlier studies, which had shown that endogenously synthesized fatty acids in cancer cells are predominantly esterified to phospholipids (which are building blocks for membranes), rather than to triglycerides as in normal tissues (53).
in vitro studies tracing the metabolic fate of [14C]-acetate in tumor cell lines (52) revealed that tumor cells incorporate 14C activity mostly into phosphatidylcholine and neutral lipids, and this incorporation showed some correlation with growth activity. Only a small fraction of 14C-acetate was converted into carbon dioxide and amino acids. This observation is in keeping with earlier studies, which had shown that endogenously synthesized fatty acids in cancer cells are predominantly esterified to phospholipids (which are building blocks for membranes), rather than to triglycerides as in normal tissues (53).
Several enzymes involved in fatty acid synthesis are up-regulated in prostate cancer, and the expression of the key enzyme fatty acid synthase (FAS) increases with increasing degree of malignancy (54). Overexpression of FAS has also been shown in bladder, lung, and endometrial cancers (55,56). FAS is a multifunctional enzyme that synthesizes palmitate from acetyl-CoA and malonyl-CoA. Under androgen stimulation, FAS expression is up-regulated in prostate cancer (57). Following androgen ablation, FAS expression initially decreases, but returns to higher levels after the transition from the androgen-responsive to the castrate-resistant state of the disease (58).
At least in prostate cancer, FAS expression appears to be regulated by the activity of the PI3K/Akt pathway. Studies have shown (a) a positive relationship between FAS and Akt expression (59), (b) an inverse relationship between PTEN and FAS expression (60), and (c) a reduction in FAS expression upon treatment with PI3K inhibitors (61). Signaling through the HER2 (ERBB2)receptor, important in a subset of breast as well as prostate cancers, has also been associated with FAS expression. High FAS expression in breast cancer is associated with a poor prognosis (62).
Although increased FAS expression is important for the progression of cancer, the reverse is also true. In breast and prostate cancer cell lines the inhibition of FAS leads to accumulation of malonyl-CoA and apoptosis (63,64). In addition to FAS, the enzyme acetyl-CoA-carboxylase α may also be critical for prostate cancer progression (65). Importantly, the newly synthesized fatty acids in cancer cells are predominantly assembled in membrane lipid rafts (66), that is, membrane microdomains that are involved in signal transduction, intracellular trafficking, cell polarization, and migration (67).
Taken together, these data indicate that fatty acid synthesis is an important metabolic process in prostate cancer progression, that at least two enzymes are critical in this process, and that inhibition or silencing of these enzymes can induce apoptosis. Acetate incorporation in cancer is a consequence of the enhanced lipid synthesis. In vitro, acetate incorporation correlates with tritium thymidine incorporation into DNA, a measure of proliferation (52). Much work remains to be done to investigate whether labeled acetate accumulation in clinical cancer studies can indeed be linked to the specific alterations in fatty acid metabolism and critical enzymes as shown in vitro and described above.
Normal biodistribution of [11C]-acetate involves intense acetate uptake in liver, spleen, and pancreas, and moderate uptake in the myocardium on early images. There is also a moderate uptake in the kidneys and varying degrees of renal excretion. Intestinal activity and uptake in skeletal muscle can vary in intensity. The effective dose equivalent is 6.2 × 10–3 mSv/MBq (68). Imaging with PET/CT is very valuable in distinguishing between bowel and urinary activity from tracer uptake in lymph nodes, bladder wall, or prostate (Table 8.18.1).
The potential pitfalls are that acetate not only accumulates in prostate and bladder cancer, but also in benign hyperplastic prostate tissue, in sites of focal prostatitis (in particular, when acute), and at sites of PIN, which may or may not progress to prostate cancer. In the prostate, acetate uptake may be age related and could be higher in individuals with normal prostate tissue who are less than 50 years of age than in normal prostate of older subjects (older than 50 years) or those with benign prostatic hyperplasia. The intensity of acetate uptake in the prostate was similar in older subjects with normal prostate (standard uptake value [SUV] 2.3 ± 0.7) and patients with proven prostate cancer (SUV 1.9 ± 0.6) (69). False positives can occur in inflammatory lymph nodes and inflammatory changes in the bladder wall.
Methionine
Amino acid uptake and protein synthesis are up-regulated in cancer. C-11-methionine is one of many possible tracers for measuring amino acid uptake in tumor cells (70). As shown in patients with castrate-resistant metastatic prostate cancer, methionine uptake in tumor occurs rapidly with a peak at about 10 minutes followed by plateau (71). Accordingly, PET imaging should start approximately 10 minutes after tracer injection. C-11-methionine undergoes rapid clearance from the blood pool, is primarily metabolized in liver and pancreas, and (in most cases) shows no significant renal excretion.
Normal biodistribution includes intense tracer uptake in the liver and pancreas and somewhat less intense uptake in spleen, kidneys, and intestines. The effective dose equivalent is 5 × 10–3 mSv/MBq (72).
Potential pitfalls include methionine uptake at sites of inflammation and misinterpretation of urinary or bowel activity as lymph node activity.
Choline
Radiolabeled choline is under investigation for imaging of prostate and bladder cancers. Studies with [14C]-choline and investigations using MR spectroscopy have established that the majority (approximately 90%) of exogenous choline is converted to phosphocholine (73,74). Phosphorylation of choline to phosphocholine is the first step of choline metabolism. Increased levels of phosphocholine in tissues is a characteristic of the malignant phenotype (74). This is likely the result of overexpression of the enzyme choline kinase, which is a common feature in many malignancies, including prostate cancer (75,76). Moreover, in breast cancer cells, the suppression of choline kinase inhibits proliferation and induces redifferentiation (77). Carbon-14 incorporation into phosphocholine—an indirect assessment of choline kinase activity—is related to the proliferative activity in various cell lines (73), although this correlation is weaker than that seen with acetate incorporation.
For PET imaging, choline has been labeled with 11C and 18F. Although [11C]-choline is an excellent agent for cancer imaging (78), the short 20-minute half-life of 11C poses a logistical challenge in many institutions. The 18F-labeled compounds include [18F]- fluoro-ethyl-choline (FEC) (79) and [18F]-fluoromethyl-dimethyl-2-hydroxyethylammonium (FCH) (80,81).
Clinically, all three labeled choline compounds show rapid clearance from blood pool and rapid uptake in prostate tissue (81,82,83), although peak uptake in prostate appears to occur at later time points with FEC than with FCH. Among the 18F-labeled choline compounds, the cellular uptake and phosphorylation by
choline kinase is very similar for FCH and natural choline, but is lower for FEC (80). In contrast to [11C]-choline, which shows very little urinary excretion and whose activity concentration in the bladder is usually lower than in prostate cancer or metastases (84), excreted 18F activity from both FCH and FEC appears in the bladder as early as 3 to 5 minutes postinjection. This may be related to incomplete tubular reabsorption of intact tracer or enhanced excretion of oxidized metabolites.
choline kinase is very similar for FCH and natural choline, but is lower for FEC (80). In contrast to [11C]-choline, which shows very little urinary excretion and whose activity concentration in the bladder is usually lower than in prostate cancer or metastases (84), excreted 18F activity from both FCH and FEC appears in the bladder as early as 3 to 5 minutes postinjection. This may be related to incomplete tubular reabsorption of intact tracer or enhanced excretion of oxidized metabolites.
Table 8.18.1 PET Tracers for Imaging of Prostate Cancer | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Excreted activity in the bladder may interfere with visualizing primary or recurrent prostate cancer. Similarly, excreted tracer in the ureter may be mistaken for uptake in nodal metastases or vice versa. Therefore, it may be helpful to start imaging of the pelvis as early as 1 minute postinjection (i.e., as soon as the majority of tracer has cleared from the blood pool). On dynamic images (e.g., 1-minute frames) true pathology will show early tracer accumulation, whereas excreted tracer in the ureters and bladder would not appear before 3 to 4 minutes after injection. Following these initial images of the lower pelvis, a PET scan of the torso, including a late image of the pelvis, is acquired. The use of combined PET/CT will aid in detecting abnormal tracer uptake in lymph nodes, in particular in smaller nodes along the course of the ureters, as well as in defining tracer accumulation in the bladder wall, prostate, or prostatic fossa after prostatectomy and distinguishing those from physiological bowel activity.
Normal biodistribution for all choline compounds includes intense uptake in liver and kidney parenchyma, and less prominent uptake in spleen, pancreas, and skeletal muscle. The effective dose equivalent is 5 × 10–3 mSv/MBq for [11C]-choline, and 3.5 × 10–2 mSv/MBq for [18F]-choline (85).
The potential pitfalls are that the intensity of choline uptake can be similar in prostate cancer, focal prostatitis, and benign prostate hyperplasia (84,86,87,88,89); SUVs as high as 5.0 were seen in benign prostatic hyperplasia. Other reasons for false-positive uptake outside of the prostate region include sarcoidosis (87),
meningiomas (87), nonspecific inflammation and infection (87), and cystitis (90). Intense bowel activity can be observed with all choline compounds and can be a reason for false-positive findings (91). False-negative findings have been described for metastases in lymph nodes of less than 1 cm in size (90,91).
meningiomas (87), nonspecific inflammation and infection (87), and cystitis (90). Intense bowel activity can be observed with all choline compounds and can be a reason for false-positive findings (91). False-negative findings have been described for metastases in lymph nodes of less than 1 cm in size (90,91).
Imaging of the Androgen Receptor in Prostate Cancer
In the normal prostate gland, androgens mediate key physiological processes such as differentiation, secretory function, metabolism, morphology, proliferation, and survival. Androgen ablation results in prostate involution and the loss of epithelial cells via apoptosis. Animal experiments have suggested that the androgen receptor (AR) functions primarily to maintain the differentiated secretory function of prostate epithelial cells, and that the survival of these cells is regulated in large part by paracrine factors expressed and provided by the supporting AR-positive surrounding stromal cells (92).
Androgens, signaling through the AR, are also the primary regulators of prostate cancer cell growth and proliferation. Androgen withdrawal causes apoptosis in a proportion of cells, whereas those that survive will arrest in the G1 phase of the cell cycle (93). Clinical progression of prostate cancer may result from continued growth of cells that were primarily resistant to androgen ablation, or from regrowth of cells that arrest after a period of growth and undergo a series of genetic alterations that permit adaptation to a low-androgen environment (94).
Primary prostate cancer and early states of recurrent disease always respond to antiandrogen hormonal therapy. Therapeutically this is used for neoadjuvant therapy in combination with external-beam radiation therapy (EBRT) (95) and in the early state of metastatic disease (96). Historically, it was assumed that progressing prostate cancer eventually reaches a hormone-refractory state where further disease progression is androgen independent. Clinical and laboratory studies in the past 5 to 10 years have shown that this concept is not true. In fact, there is continued signaling through the AR in progressive prostate cancer despite castrate serum levels of testosterone, and this phenomenon continues into the late states of the disease (97,98,99). Intratumoral androgens are also found even in late stages of the disease.
Exactly why and how even advanced stages of the disease remain dependent on signaling through the AR is still under investigation. Recent experimental studies have shed some light into this. According to classic teaching, steroid hormones exert most of their effects by binding to specific nuclear receptors, which then act as transcription factors. For many years, such “genomic function” was also thought to be the main or only function of the androgen receptor: androgen binding to the AR S AR translocation to the nucleus S binding to androgen responsive elements on the DNA S stimulation of mRNA synthesis S protein synthesis.
It has been suggested that the progression from low-grade to high-grade prostate cancer and metastases is mediated by a selective down-regulation of those AR target genes that inhibit proliferation, induce differentiation, and mediate apoptosis, whereas target genes that promote the growth and survival of prostate cancer cells are still expressed (100). However, it is now clear that in addition to its genomic function the AR can also act directly as a cytoplasmic signaling molecule. For instance, signaling through the AR can stimulate the PI3K/Akt pathway through direct interaction of the AR with the p85α regulatory subunit of PI3K (101,102), thus protecting cells against apoptosis and promoting tumor proliferation.
In addition, stimulation of the AR by binding of its natural ligand dihydrotestosterone can cause direct activation of mTOR, which does not require activation of the PI3K/Akt or MAP (mitogen activated protein) kinase pathways (103). Evidence suggests that mTOR activation in this setting is responsible for the increased expression of multiple genes whose protein products are involved in enhancing the availability of nutrients in the cancer cell.
The various mechanisms explaining continued signaling through the AR in advanced prostate cancer include receptor activation by ligands other than testosterone (including AR antagonists), a lowering in the threshold for ligand necessary for the receptor activation, increased expression of steroid receptor coactivators, and many more (98,104). For instance, studies in mice have shown that signaling through the Her2/Her3 receptor dimer stabilizes the AR and promotes its binding to target genes on the DNA (105). This is one example for how signaling through the AR does not necessarily require the presence of androgens for receptor activation. Other in vivo studies have demonstrated crosstalk between the AR and the epidermal growth factor receptor (EGFR) in prostate cancer cells. A functional AR appeared important in mediating the signaling through EGFR (106), again regardless of the presence or absence of androgens.
At the cellular level, a direct synergy between AR and Akt signaling can transform normal prostate epithelial cells and induce androgen insensitive carcinoma. Although these cells continue to proliferate despite androgen withdrawal, they do require an AR with maintained genomic and nongenomic function (see above). Akt seems to regulate the expression and stability of AR at multiple levels (107). Finally, hypoxia can also affect the function of the AR by stimulating the binding of the AR to the androgen-responsive element on the DNA and enhancing the activity of genes that are expressed under the control of the AR (108).
As mentioned before, the AR is expressed on both prostate epithelial as well as surrounding stromal cells. Most recent studies indicate that hormone-refractory prostate cancer is characterized by a stimulation of AR function on epithelial cells (thus promoting proliferation) and simultaneous blockage of AR function on surrounding stromal cells (thus blocking apoptosis-inducing paracrine signals from the stroma). This alteration in regional AR expression within prostate tissue appears to be a consequence of an activation of the PI3K/AKT pathway (and perhaps also the Erk/MAP kinase pathway) (109).
Because continued signaling through the AR occurs even in advanced prostate cancer and in fact promotes disease progression, pharmacologic studies are now under way to target the AR and cause its destruction or down-regulation. Experimental studies using HSP-90 inhibitors (110), RNA interference (111), or short hairpin RNA (112) have provided proof of concept in this regard. (HSP-90 is a “chaperone” protein needed for the stabilization and function of the androgen receptor.) It is therefore of increasing interest to develop a method to measure AR expression and activity. This should improve our understanding of prostate cancer biology and should be helpful in monitoring the response to drugs targeting this receptor. Potentially, such method could also identify candidates more likely to respond to certain classes of drugs or small molecules targeting the androgen receptor.
Verification of the AR expression in patients with metastatic prostate cancer would require repeated biopsies from multiple sites.
A noninvasive method for systemic and repeated study of AR expression and function would therefore be preferred. Several compounds were developed for this purpose (113), and the agent 16β-[18F]-fluoro-5α-dihydrotestosteron (FDHT) is currently under clinical investigation for imaging and quantifying the androgen receptor expression in metastatic prostate cancer (114,115).
A noninvasive method for systemic and repeated study of AR expression and function would therefore be preferred. Several compounds were developed for this purpose (113), and the agent 16β-[18F]-fluoro-5α-dihydrotestosteron (FDHT) is currently under clinical investigation for imaging and quantifying the androgen receptor expression in metastatic prostate cancer (114,115).
Larson et al. (114) studied the biodistribution and binding characteristics of FDHT. Among seven patients imaged with both FDG and FDHT, 46 of 59 lesions showed both FDG and FDHT uptake, and the remainder had FDG but not FDHT uptake. The normal biodistribution of this agent involves high uptake in liver with excretion through the hepatobiliary system, causing some bowel activity, uptake in kidneys with some excretion to bladder, and high blood pool activity in the heart and large arteries. The effective dose equivalent is 1.8 × 10–2 mSv/MBq, which is similar to FDG (116). FDHT is a suboptimal radiotracer because of its high lipophilicity, rapid metabolism, and the persistent high blood pool activity of breakdown products. Newer PET tracers for imaging the AR are therefore under development.
Tumor Proliferation
Fluorine-18-fluorothymidine (FLT) (117) is a promising proliferation marker for PET imaging studies, but there are currently no conclusive data on FLT imaging in patients with urologic cancers. In a mouse model using the CWR22 androgen-dependent xenograft, FLT uptake in tumor reached its plateau in 30 minutes and remained up to 60 minutes. Surgical castration or treatment with diethylstilbestrol caused a marked reduction of FLT uptake in the tumor (118). Because of high natural uptake in bone marrow, the agent may not be suitable for the imaging of osseous metastases. Whether FLT may be useful for treatment monitoring in metastatic renal or prostate cancer or for detecting viable cancer in residual nodal masses after chemotherapy for testicular cancer, remains to be seen, but concerns exist as to whether the degree of tumor targeting will be sufficient.
Summary and Recommendations
Imaging genitourinary cancers with PET poses several challenges. Although FDG is a useful tracer, it is clearly limited in several settings, and alternative tracers are necessary for optimum diagnosis. Given the rapid evolution of this field, the optimal diagnostic tracer is not yet fully clarified in several settings. Regardless, for each radiotracer employed, the interpreting physician needs to understand the normal biodistribution and normal variants in tracer accumulation. None of the available radiotracers is specific for cancer, and false-positive tracer uptake (in benign lesions or normal anatomic structures) can occur for many reasons. However, depending on the underlying disease, the location of a hypermetabolic lymph node might determine the probability for a metastasis. For instance, inguinal lymph nodes are an extremely unusual site for metastases from prostate or bladder cancer, whereas nonspecific enlargement and tracer uptake in inguinal nodes is a relatively common finding in many patients. It is helpful to establish a threshold for the intensity of tracer uptake that should be considered normal or abnormal in order to guarantee the reproducibility of study interpretation and standardize the reporting. Depending on the radiotracer, this could include a comparison to uptake in surrounding background, uptake in large blood vessels, or uptake in skeletal muscle. Measurements of the SUV may guide the interpretation in a given case, but they cannot be used categorically for differentiating between cancer versus normal tissue, inflammation or infection.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


