Head and Neck
Amy F. Juliano
Sara O. Vargas
Caroline D. Robson
INTRODUCTION
Anatomically, “head and neck” encompasses structures from the skull base to the thoracic inlet: the orbits, face, sinuses and nasal cavity, bony skull base, temporal bones, soft tissues of the neck, and upper aerodigestive tract including the pharynx and larynx. Various congenital and acquired anomalies and abnormalities arise from these structures, and imaging evaluation plays an important role for initial diagnosis and follow-up assessment. In this chapter, imaging techniques for evaluating head and neck in pediatric patients are discussed, and normal anatomy is reviewed. In addition, various commonly and occasionally encountered disorders affecting the pediatric head and neck are presented, including clinical features, characteristic imaging findings, relevant pathologic findings, and current treatment approaches.
IMAGING TECHNIQUES
Radiography
Trauma and inflammatory disease in the sinonasal and orbital regions are sometimes initially evaluated by radiographs. Frontal, lateral, and Waters projections are useful. When obtained in the upright position, air-fluid levels can be appreciated. For mandibular trauma, AP, Towne, oblique, and Panorex views are obtained. Radiographs are also useful when a radiopaque foreign body is suspected. Orbital views are sometimes obtained prior to performing magnetic resonance imaging (MRI) to exclude the presence of a metallic foreign body as a potential contraindication to MRI.
Radiographs are valuable in the assessment of pediatric patients following cochlear implantation. The AP transorbital projection allows visualization of the entire length of the internal auditory canal (IAC) and is useful for confirming appropriate electrode position.1 A modified Stenvers view (PA oblique with the midsagittal plane forming an angle of 50 degrees with the plane of the film and the central ray 2 cm above and parallel to the horizontal plane) allows visualization of the entire intracochlear electrode array (Fig. 3.1)2 and can ensure correct positioning and absence of kinking.3 Postoperative radiographs are recommended when intraoperative radiographs cannot confirm electrode placement, when extrusion is suspected, and for evaluating nonauditory responses.4
Lateral radiographs of the neck are valuable for evaluating upper airway abnormalities such as adenoidal hypertrophy, retropharyngeal swelling, epiglottitis, and airway stenosis (Fig. 3.2). Frontal views of the neck are indicated for foreign body ingestion/inhalation, and mass effect on the larynx and upper trachea.5,6,7,8,9
Ultrasound
Real-time ultrasound (US) is useful for determining the size, location, and characteristics of lesions in the head and neck.6,9,10,11,12 Ultrasound differentiates solid from cystic lesions and identifies nodal architecture distinguishing nodes from nonnodal masses. Calcification appears hyperechoic with posterior acoustic shadowing, and fat appears moderately hyperechoic. If a cystic structure with echogenic contents is suspected, it is useful to ballot the mass to detect the swirling motion of echogenic particles within. Doppler US provides information about blood flow. US also provides guidance for lesion biopsy, abscess drainage, and injections such as botulinum toxin A.
 FIGURE 3.1 Modified Stenvers radiographic view of cochlear implant electrode. Landmarks include the internal auditory canal (arrows), superior semicircular canal (large arrowhead), and lateral semicircular canal (small arrowhead). The cochlea is located anterior to the semicircular canals and inferior to the internal auditory canal. The tip of the electrode can be seen to form a 360-degree turn in the expected location of the cochlea. |
Computed Tomography
Computed tomography (CT) is the primary imaging modality for evaluation of bony architecture and acute inflammatory processes in the head and neck.6,9,10,12,13,14,15 CT provides excellent delineation of soft tissue structures against a background of fat, bony structures against a background of air or fluid; detects calcifications (e.g., phleboliths, sialoliths, and tumoral calcification); and shows bony remodeling or erosion. Contrast-enhanced neck CT is generally obtained using a split bolus technique in which the scout is obtained and the first half of the IV bolus is injected, followed by a 3-minute wait, and then acquisition of helical images during the rapid injection of the second half of the bolus. This technique allows assessment of vessels, abnormal vascularity, and abnormal permeability from inflammation or tumor. The excellent spatial resolution of CT is useful for evaluating small congenital anomalies such as branchial sinus tracts.
 FIGURE 3.2 Lateral radiograph of the neck in an 18-month-old girl who presented with croupy cough and biphasic stridor. Radiograph demonstrates severe subglottic stenosis (white arrow). A subglottic mass was noted and biopsied during direct laryngoscopy. The mass proved to be ectopic thymic tissue. The child also has adenoidal prominence, focally contacting the soft palate (black arrowhead). |
For evaluation of small anatomic structures, multiplanar reformatted CT images are helpful. Three-dimensional (3D) CT reconstructions are useful in evaluating traumatic injuries and craniofacial malformations for surgical planning. CT angiography and venography provide details of the arteries and veins of the head and neck for a variety of underlying vascular anomalies and abnormalities
Intraocular lesions are usually evaluated via clinical ophthalmologic assessment and US. CT is mainly used for detection of foreign bodies and intraocular calcification,9 and in the setting of trauma. For intraocular tumors and other intraocular pathology, MRI is the preferred initial imaging study.16,17,18,19,20 With increasing awareness and concern regarding the effects of radiation in pediatric patients with hereditary retinoblastoma, in whom there is higher risk of developing second primary malignancies,21 MRI has now been established as the imaging modality for diagnosis, staging, and treatment monitoring.22
Nonenhanced CT is used to evaluate facial and orbital trauma and for preoperative assessment of chronic sinus disease, often providing information that can be used with intraoperative guidance systems. Axial thin sections are obtained, with submillimeter multiplanar bone algorithm reformats. For acute complicated sinonasal and orbital infection and nonocular tumors, contrast-enhanced CT precisely delineates osseous structures. Evaluation of dental lesions and the alveolar processes of the maxilla and mandible is primarily accomplished by CT, increasingly with the use of low-dose cone beam units.
In the temporal bone region, CT is the preferred initial modality for evaluation of congenital external ear and middle ear malformations, conductive hearing loss, suspected retrotympanic mass, suspected jugular dehiscence, vascular variants such as a persistent stapedial artery, and inner ear malformations.9,10,12,13,14 Infection and inflammation, including cholesteatoma, and bone destruction are also best evaluated by CT. Suspected coalescent mastoiditis and tumors require the use of intravenous contrast in order to detect intracranial involvement and dural venous sinus thrombosis.
Magnetic Resonance Imaging
MRI is complementary to CT in evaluating head and neck pathology and is sometimes the imaging modality of choice. In general, MRI is a superior technique for delineation of
soft tissue elements, vascular components, and intracranial involvement. Skull base evaluation requires a combination of CT for bony involvement and MRI for neurovascular, soft tissue, and marrow involvement. MR angiography (MRA) and MR venography (MRV), Doppler US, and CT angiography are additional techniques helpful for assessment of vascular flow, vessel caliber, and vascular tumors and malformations. Indications for temporal bone MRI include sensorineural hearing loss (SNHL), complicated coalescent mastoiditis, and facial nerve paralysis.
soft tissue elements, vascular components, and intracranial involvement. Skull base evaluation requires a combination of CT for bony involvement and MRI for neurovascular, soft tissue, and marrow involvement. MR angiography (MRA) and MR venography (MRV), Doppler US, and CT angiography are additional techniques helpful for assessment of vascular flow, vessel caliber, and vascular tumors and malformations. Indications for temporal bone MRI include sensorineural hearing loss (SNHL), complicated coalescent mastoiditis, and facial nerve paralysis.
Commonly used MRI techniques for head and neck imaging include fast spin echo (FSE) T1- and T2-weighted imaging, inversion recovery (IR) sequences, fat-suppressed sequences, heavily T2-weighted sequences, diffusion-weighted imaging, flow-sensitive sequences, T1-weighted non-fat-suppressed sequences, and T1-weighted, fat-suppressed, contrast-enhanced sequences. Imaging at 3T with the use of thin-section 3D heavily T2-weighted sequences with submillimeter slice thickness is used for assessment of the membranous labyrinth of the inner ear, cranial nerves, and ocular abnormalities.
Fluoroscopy
Fluoroscopy provides dynamic information not available with radiographs. Video fluoroscopy in conjunction with still images is useful in the assessment of a child with stridor, difficulty feeding, or chronic airway obstruction. Airway caliber and contour in various phases of respiration can be observed, and an airway mass, laryngomalacia, or tracheomalacia may be detected. When performed in conjunction with a barium pharyngogram/esophagogram, vascular rings or slings, mediastinal masses, or esophageal foreign bodies or masses may be detected as well.9
Barium pharyngogram may demonstrate pyriform sinus tracts. The exam is performed after resolution of neck inflammation, to reduce false-negative rate related to effacement of the fistulous tract by edema.
Nuclear Medicine
Nuclear medicine is most useful in the head and neck for thyroid gland evaluation. Iodine-123 (I-123) and technetium-99m (Tc-99m) pertechnetate are the main agents used. I-123 is trapped and organified by the thyroid, whereas Tc-99m pertechnetate is trapped but not organified. Common indications for thyroid scintigraphy include identification of ectopic thyroid tissue and evaluation of thyroid nodules.
Octreotide scans may be helpful for identifying paragangliomas, especially in the setting of familial syndromes where there may be multiple tumors. Fluorodeoxyglucose (FDG)-positron emission tomography (PET) CT is used to aid in the diagnosis and staging of head and neck malignancies.
ORBIT AND GLOBE
Embryology and Development
The eye (globe) develops from the neuroectoderm of the forebrain, the surface ectoderm of the head, the mesoderm between these two layers, and neural crest cells.9,23 The neuroectoderm gives rise to the retina, optic nerve, and smooth muscles of the iris. The overlying surface ectoderm gives rise to the corneal epithelium, conjunctival epithelium, lens, and lacrimal and tarsal glands. The mesenchyme and neural crest cells give rise to the corneal stroma, sclera, choroid, iris, ciliary musculature, part of the vitreous body, and cells lining the anterior chamber.24
The optic primordium gives rise to the optic vesicle. The overlying surface ectoderm thickens and forms the lens placode. The outer surface of the optic vesicle then flattens and becomes concave, resulting in the optic cup, whereas the proximal portion of the optic vesicle constricts and forms the optic stalk.23,24
In the 5 mm embryo, invagination occurs along the optic stalk, forming a groove. At the distal end, the groove opens into the optic fissure or choroidal fissure, at the inferior edge of the optic cup. Through this fissure, mesenchyme extends into the optic stalk and cup, carrying with it the hyaloid artery, a branch of the ophthalmic artery. Over time, the edges of the fissure approximate and the groove narrows, until the fissure closes and the groove becomes the optic canal, inside the optic stalk. Failure of the fissure to close completely results in a coloboma, which may be seen anywhere along the length of the optic stalk and can affect any of the optic stalk derivatives.
The hyaloid artery travels through the globe within the hyaloid canal during development of the primitive or primary vitreous. Over time, the embryonic hyaloid vascular system involutes. The hyaloid artery and vein become the central artery and vein of the retina, coursing within and along the optic nerve. Persistence and hyperplasia of the embryonic hyaloid system in the globe result in persistent hyperplastic primary vitreous (PHPV).9
Normal Anatomy
The globe is embedded in orbital fat and is surrounded by a thin capsule.9,25,26,27 The opaque sclera and the transparent cornea form the outer layer of the globe. The middle layer is formed by the uvea, which is vascular and contains pigmented tissue including the choroid, ciliary body, and iris. The retina, the inner layer, is the neural, sensory layer that receives images. The retina is continuous posteriorly with the optic nerve. The refracting media of the eye are the aqueous humor, the crystalline lens, and the vitreous humor. The lacrimal gland is situated in the superolateral orbit. The puncta of the superior and inferior canaliculi are located in the medial aspect of the upper and lower lids. They travel through the superior and inferior canaliculi to the common canaliculus and then enter a small diverticulum of the lateral wall of the lacrimal sac, called the sinus of Maier, and then into the lacrimal sac proper.9 The nasolacrimal duct (NLD) extends from the lacrimal sac and empties into the inferior meatus of the nasal cavity.
The orbit contains the orbital fascia, extraocular muscles, globe and its appendages, vessels, nerves, and fat.9,25,26,27 The optic foramen lies at the orbital apex and transmits the optic nerve and ophthalmic artery. The superior orbital fissure lies inferolateral to the optic foramen and transmits cranial nerves III and IV, the ophthalmic division of cranial nerve V, cranial nerve VI, sympathetic nerves, and the ophthalmic vein. The orbital floor and lateral orbital wall are separated posteriorly by the inferior orbital fissure. This fissure connects the orbit with the pterygopalatine fossa posteromedially and with the retromaxillary fissure and infratemporal fossa anterolaterally.
Except for the inferior oblique, the extraocular muscles originate at the orbital apex and insert on the globe. The inferior oblique muscle arises from the orbital floor just lateral to the opening of the nasolacrimal canal. The orbital fascia forms the periosteum of the orbit. Anteriorly, it forms a circumferential reflection known as the orbital septum. Structures anterior to the septum are termed preseptal; those posterior to it are termed postseptal. The orbital septum provides a barrier to the posterior spread of preseptal inflammation. The postseptal space may be further subdivided by the cone of muscles into intraconal and extraconal compartments.
Congenital and Developmental Anomalies
Malformations of the Globe
Anophthalmia
Anophthalmia is congenital absence of the eye, a rare unilateral or bilateral abnormality. By definition, true anophthalmia characterized by complete absence of ocular tissue within the orbit is rare as there is usually a small eye or a dysplastic remnant present. Anophthalmia is associated with infections and congenital syndromes such as trisomy 13 and complex craniofacial malformations.9,26,27,28,29
Primary anophthalmia results from failure of formation of the optic vesicle. Consecutive anophthalmia is when the optic vesicle forms but then degenerates. Secondary anophthalmia and congenital cystic eyeball are caused by partial or complete failure of the primary optic vesicle to involute early in development. When the cyst is small or nonexistent, anophthalmia results. When the cyst is large, a congenital cystic eyeball results.30
Microphthalmia
In microphthalmia, a small globe and a lens are present, although severe microphthalmia may be difficult to distinguish from anophthalmia clinically and radiologically (Fig. 3.4).30 A microphthalmic eye that otherwise demonstrates normal architecture is called a nanophthalmic eye. Microphthalmia may exist alone or may be associated with other abnormalities such as glaucoma, cataract, genetic syndromes (e.g., trisomy 13 and holoprosencephaly), and TORCH infections. Microphthalmia with a retrobulbar duplication cyst occurs when ocular tissue prolapses through a coloboma defect (persistent embryonic optic fissure).
 FIGURE 3.3 Anophthalmia in an 8-month-old girl. Axial T2-weighted fat-suppressed MR image shows absence of globes with shallow orbits bilaterally. A small cyst on the left may represent dysplastic remnant (arrow). The child has SOX2 anophthalmia syndrome, having esophageal atresia and cryptorchidism as well. |
Macrophthalmia
A number of conditions can lead to enlargement of the globe. The most common condition is pathologic axial myopia, where there is elongation of the globe because of increased anteroposterior dimension of the posterior chamber. Macrophthalmia is sporadic or syndromic in nature (e.g., Stickler syndrome) and may be asymmetric (Fig. 3.5). The abnormal elongation leads to increased risk of retinal abnormalities and staphylomas.
 FIGURE 3.4 Microphthalmia. Axial CT image demonstrates a very small left globe (arrow) and normal-sized right globe. |
 FIGURE 3.5 Macrophthalmia. Axial T2-weighted fat-suppressed MR image shows an enlarged right globe. This 14-month-old boy has a developmental defect in the retina and optic nerve with subnormal vision. His poor vision is thought to be related to amblyopia that is a result of restricted movement of the abnormally elongated globe. He does not have elevated intraocular pressure, and therefore this does not represent buphthalmos. |
In buphthalmos, the globe is enlarged because of increased intraocular pressure at a time when the sclera can be stretched beyond its normal shape and size, as it is prior to age 4 years. There is increased anteroposterior dimension of both the anterior and posterior chambers. The increase in intraocular pressure may be because of congenital primary glaucoma or because of glaucoma related to an underlying condition such as aniridia, neurofibromatosis, or Sturge-Weber syndrome.
Staphyloma
Staphyloma is characterized by thinning and stretching of the scleral-uveal coats of the globe. All the layers are present, and there is no defect in the layers as in coloboma. It usually occurs posteriorly (Fig. 3.6); anterior (corneal) staphyloma is rare. Affected pediatric patients typically present with severe axial myopia. Posterior staphyloma may be seen in high axial myopia, glaucoma, scleritis, and necrotizing infection, or may be from iatrogenic causes. Anterior staphyloma is seen secondary to inflammation involving the scleral-corneal layer.9
 FIGURE 3.6 Posterior staphyloma. Axial CT image demonstrates thinning and stretching of the posterior surface of the globe on the left, resulting in elongation of the globe. |
Coloboma
A coloboma results from incomplete closure of the embryonic optic fissure. A coloboma can involve the cornea, iris, lens, retina, choroid, sclera, and optic nerve, in a continuous or discontinuous fashion, in the inferior aspect. Unlike staphyloma where the uveal-scleral layers are thinned but present, in coloboma, there is a complete structural defect, with a gap through the involved structure(s). Half of all typical colobomas are bilateral.31 Coloboma is seen as a defect at the posterior globe with focal outpouching of the globe contour at the level of the optic nerve insertion (Fig. 3.7).
In some cases, there may be herniation of dysplastic tissue through the defect.32,33 A cyst may result, sometimes communicating with the globe, which is small and dysplastic. This is termed microphthalmia with cyst or a colobomatous cyst (Fig. 3.8). The cyst is often larger than the globe, and the cyst content is more similar to cerebrospinal fluid (CSF) than to vitreous.34 Colobomatous cysts are associated with systemic syndromes such as CHARGE syndrome, VATER association, Warburg syndrome, and trisomies 13 and 18.
Morning Glory Disc Anomaly
Morning glory disc anomaly (MGDA) is a congenital dysplasia of the optic disc/optic nerve, with a funnel-shaped excavation at the optic disc. The appearance resembles a morning glory flower, hence the name. On funduscopic examination, an annulus of chorioretinal pigmentary change surrounds the optic disc excavation, and a central glial tuft overlies the optic disc.35,36 The condition is usually unilateral and
sporadic, although there is an association with other ocular and intracranial abnormalities such as midline craniofacial defects, callosal dysgenesis, and basal encephaloceles; vascular abnormalities including segmental aplasia of the circle of Willis and moyamoya disease as seen in PHACES association; and other cerebral malformations.35,37 Therefore, pediatric patients with MGDA should undergo brain MRI and MRA as part of their imaging workup. To date, the pathogenesis of MGDA is unknown.
sporadic, although there is an association with other ocular and intracranial abnormalities such as midline craniofacial defects, callosal dysgenesis, and basal encephaloceles; vascular abnormalities including segmental aplasia of the circle of Willis and moyamoya disease as seen in PHACES association; and other cerebral malformations.35,37 Therefore, pediatric patients with MGDA should undergo brain MRI and MRA as part of their imaging workup. To date, the pathogenesis of MGDA is unknown.
 FIGURE 3.7 Colobomas. Axial T2-weighted fat-suppressed MR image shows a defect at the posterior right globe at the site of the optic nerve insertion, with a focal outpouching at that location. This is also present on the left (arrows). |
 FIGURE 3.8 Colobomatous cyst in a 3-year-old girl. Axial CT image demonstrates a colobomatous cyst (arrow) posterior to the right globe. The globe is smaller than normal in size and dysplastic (arrowhead). |
On imaging, a funnel-shaped morphology of the optic disc can be seen (Fig. 3.9). In contradistinction to coloboma, elevation of the retinal margins adjacent to the excavation is characteristic. On MRI, one may appreciate abnormal T1 hyperintense soft tissue associated with the distal portion of the optic nerve, representing the glial tissue seen on ophthalmic examination. The ipsilateral optic nerve is sometimes atrophied.36
Malformations of the Orbit
Hypertelorism and Hypotelorism
Hypertelorism refers to an increased distance between the medial walls of the orbits.5,6,7,8,9 This is to be distinguished from telecanthus, where the distance between the apices of the medial canthal ligaments is increased and the eyes appear to be farther apart than normal but the bony interorbital distance is not increased. A combination of telecanthus with lateral displacement of the lacrimal puncta, known as dystopia canthorum, is characteristic of some types of Waardenburg syndrome.38 Causes of hypertelorism include familial hypertelorism, median cleft face syndrome, cleidocranial dysplasia, syndromic coronal craniosynostosis, cephalocele, trisomy 13, and Hurler disease.
Hypotelorism refers to a bony interorbital distance that is less than normal. Underlying causes of hypotelorism include arhinencephaly, holoprosencephaly, microcephaly, trigonocephaly, sagittal craniosynostosis, and primary developmental abnormalities of the eyes.
 FIGURE 3.9 Morning glory disc anomaly in a 4-year-old girl. Axial T2-weighted SPACE MR image shows funnel-shaped morphology of the posterior left globe at the level of the optic disc. Elevated retinal margin can be seen laterally (arrow). |
Large or Small Orbit
A large orbit may be caused by brain herniation through a mesenchymal bony defect (greater sphenoid wing in neurofibromatosis), cephalocele, or an increase in orbital contents present early in development from, for example, neurofibroma or vascular anomaly.5,6,7,8,9
A small shallow orbit is seen in congenital anophthalmia, in microphthalmia, and as a sequela of early insult, for example orbital enucleation and radiation. Exorbitism refers to protrusion of the globe due to a shallow orbit and is characteristic of some forms of syndromic craniosynostosis, or may occur from hyperostotic changes in the orbit as seen in fibrous dysplasia (Fig. 3.10) and osteopetrosis.
 FIGURE 3.10 Fibrous dysplasia of the sphenoid wing causing narrowing of the optic canal in an 11-year-old girl who presented with left proptosis. Axial T2-weighted fat-suppressed MR image demonstrates expansion of the greater wing of the sphenoid on the left, including the anterior clinoid process, due to fibrous dysplasia. The left orbit is smaller than normal, and the optic canal is narrowed (arrow). |
 FIGURE 3.11 Optic nerve glioma in neurofibromatosis type 1. Axial postcontrast T1-weighted fat-suppressed MR image shows a fusiform enhancing mass along the left optic nerve representing the optic nerve glioma. The left optic canal is expanded as a result (arrows). |
Large or Small Optic Canal
A large optic canal is almost always due to an intracanalicular optic glioma (Fig. 3.11). Rare causes include other tumors (e.g., neurofibroma, meningioma, hemangioma) and dural ectasia (e.g., in neurofibromatosis). A small optic canal is due to a small optic nerve (e.g., in microphthalmia, optic nerve hypoplasia or atrophy, early orbital enucleation) or bony hyperostosis (e.g., fibrous dysplasia, osteopetrosis) (Fig. 3.10).
Optic Nerve Hypoplasia
Optic nerve hypoplasia, a subnormal number of axons,26,27,28,29 is a frequent anomaly and is often isolated. Particularly when bilateral, it may be associated with ocular, facial, endocrine, or central nervous system (CNS) anomalies, among which are septooptic dysplasia and encephalocele. Although it is primarily a clinical diagnosis, characteristic MR findings are small optic nerves and a small chiasm.
 FIGURE 3.12 Persistent Hyperplastic Primary Vitreous (PHPV) in a 47-day-old infant. A: Axial CT image shows left microphthalmia, with increased density in the vitreous chamber (arrow). B: Axial postcontrast T1-weighted fat-suppressed MR image shows T1 hyperintense signal in the vitreous chamber (black arrow) and a retrolental mass with enhancing areas (white arrow). The Cloquet canal can be seen (black arrowhead). |
TABLE 3.1 Differential Diagnosis of Leukocoria | ||||
|---|---|---|---|---|
|
Primary Ocular Abnormalities
Persistent Hyperplastic Primary Vitreous
PHPV is caused by failure of the embryonic hyaloid vascular system to regress, resulting in persistence and hyperplasia of the primary vitreous, tunica vasculosa lentis (capillary vascular network covering parts of the lens), and associated embryonic connective tissue. PHPV is usually unilateral and associated with microphthalmia. The typical presentation is unilateral leukocoria (white pupil) with microphthalmia and cataract in a term infant. After retinoblastoma, PHPV is the most common cause of leukocoria in childhood (Table 3.1).39 The two entities may be differentiated on CT by the presence of microphthalmia and absence of calcification in PHPV. Retinoblastoma usually contains calcifications and involves a normal or large globe. When glaucoma complicates PHPV, it leads to buphthalmos. Other complications include recurrent hemorrhage, retinal detachment, and phthisis bulbi.
On CT, there is increased density of the vitreous chamber (Fig. 3.12A) and enhancement of abnormal intravitreal tissue. Intravitreal densities suggest the persistence of fetal tissue in the Cloquet canal or congenital nonattachment of the retina. Microphthalmia ranges from obvious to subtle, and calcification is absent within or around the globe. The lens may be small and irregular, and the anterior chamber may be shallow.
On MR, there is T1 hyperintensity of the vitreous chamber and enhancement of abnormal intravitreal tissue (Fig. 3.12B).
A discrete linear structure suggestive of the Cloquet canal may be seen. An enhancing retrolental mass may be present. The anterior form of PHPV is characterized by flattening of the lens, a shallow anterior chamber, and enhancement of the anterior segment structures. PHPV is often associated with optic nerve and retinal malformations.40
A discrete linear structure suggestive of the Cloquet canal may be seen. An enhancing retrolental mass may be present. The anterior form of PHPV is characterized by flattening of the lens, a shallow anterior chamber, and enhancement of the anterior segment structures. PHPV is often associated with optic nerve and retinal malformations.40
 FIGURE 3.13 Norrie disease. Axial postcontrast T1-weighted fat-suppressed MR image demonstrates bilateral microphthalmia. The vitreous is T1 hyperintense in signal, and there are retrolental masses (arrows). |
Norrie Disease
Norrie disease, or congenital progressive oculoacousticocerebral degeneration, is a rare, X-linked, recessive disorder consisting of retinal malformation, deafness, and mental retardation or deterioration. Early-onset blindness is due to severe retinal dysplasia. Clinical findings include leukocoria, iris atrophy, retrolental fibroplasia, vitreous hemorrhage, retinal dysplasia, retinal folds, and retinal detachments.41 The end result is cataracts and opaque corneas, and, eventually, phthisis bulbi.
On imaging, the vitreous chambers are dense on CT and T1 hyperintense on MRI bilaterally (Fig. 3.13). A retrolental mass, retinal detachment, shallow anterior chamber, small dense lens, and optic nerve atrophy and microphthalmia may be seen.9
 FIGURE 3.14 Coats disease. Axial T1-weighted (A) and T2-weighted fat-suppressed (B) MR images demonstrate a large subretinal exudate occupying most of the vitreous chamber of the left globe, hyperintense to white matter on both sequences (arrows). |
Coats Disease
Coats disease (primary retinal telangiectasias) is a primary retinal vascular anomaly characterized by telangiectatic and aneurysmal retinal vessels, and the accumulation of lipoproteinaceous exudate in the retina and subretinal space, leading to massive exudative retinal detachment.26,27,28,29 Leukocoria may be seen with the occurrence of retinal detachment. The peak incidence is near the end of the first decade, with a higher incidence in males.
Early in the disease, imaging findings may be minimal. Subsequent findings are largely related to retinal detachment. The subretinal exudate of Coats disease is usually hyperintense on T1- and T2-weighted MR sequences (Fig. 3.14) and hyperdense on CT. Coats disease can simulate retinoblastoma, with a mass that should be heterogeneous in density and signal and nonenhancing. Calcification sometimes occurs.
Retinopathy of Prematurity
Retinopathy of prematurity (ROP) (retrolental fibroplasia) is seen in premature low-birth-weight infants who receive prolonged oxygen therapy. Currently, the prevalence of the abnormality has decreased because of judicious use of oxygen in infants.
It is usually bilateral, and may be symmetric or asymmetric. The pathophysiology is not completely understood; excessive oxygen has been implicated. There is proliferation of abnormal peripheral retinal vessels, with subsequent hemorrhage and cicatrization. In the late stage, a dense membrane or vascularized mass is left with tractional retinal detachment and microphthalmos.9
In more advanced cases, it may be difficult to differentiate ROP from PHPV and other conditions resulting in microphthalmia with associated retinal detachment.42,43 A history of prematurity, incubator treatment, low birth weight, bilaterality, and microphthalmia helps confirm the diagnosis of ROP. Calcification is seen in advanced cases.
Congenital Glaucoma
Glaucoma is abnormal elevation of intraocular pressure,26,27,28,29 usually caused by increased resistance to normal outflow of aqueous humor. The increase in intraocular pressure at a young age, when the sclera is still malleable, leads to buphthalmos. Hydrophthalmos refers to the high fluid content
present with marked enlargement of the eye that is seen in any type of glaucoma present since infancy.44
present with marked enlargement of the eye that is seen in any type of glaucoma present since infancy.44
Ocular and Orbital Abnormalities Associated with CNS Malformations
Disorders of neural tube closure in which there may be orbital abnormalities include cephaloceles, dermal sinus and cyst, neuroglial heterotopia, holoprosencephaly, septooptic dysplasia, absence of the septum pellucidum, craniosynostosis, and cranial facial syndromes.45 Cephaloceles that commonly involve the orbit or optic pathways include sphenoidal, nasoorbital, and frontoethmoidal cephaloceles.25,46,47,48,49,50,51 Dermal sinuses, dermoids, and epidermoids are discussed later. Widening of the nasal bridge and hypertelorism should prompt a search for craniofacial anomalies, cephaloceles, and midline intracranial defects such as callosal agenesis.
Midface hypoplasia and hypotelorism are commonly associated with the holoprosencephalies.45,47,52 Midface and orbital anomalies associated with alobar holoprosencephaly include cyclopia (single midline orbit with proboscis and absent nose), ethmocephaly (median proboscis between two hypoteloric orbits), cebocephaly (rudimentary nose with single aperture and orbital hypotelorism), the rare median cleft lip with hypertelorism, and simple hypotelorism. Septooptic dysplasia anomalies with deficiency of the septum pellucidum and optic hypoplasia are considered a mild form of holoprosencephaly.45,52,53
Unilaterally or bilaterally deformed orbits are commonly associated with craniosynostoses, particularly those involving the metopic suture, coronal suture, or multiple sutures.45,50 Hypertelorism with or without exorbitism is characteristic of syndromic coronal synostosis. Reconstructive craniofacial surgery is often required to improve appearance and preserve vision. Metopic synostosis results in hypotelorism. Treacher Collins syndrome (TCS) (mandibulofacial dysostosis) is as associated with microphthalmia and coloboma.
Neuroophthalmologic involvement often occurs with the neurocutaneous syndromes of childhood such as neurofibromatosis type 1 (NF1) (sphenoorbital dysplasia [Fig. 3.15]), optic glioma (Fig. 3.11), tuberous sclerosis (retinal neuroglial hamartoma), Sturge-Weber syndrome (choroidal venocapillary malformation with buphthalmos), and von Hippel-Lindau disease (retinal hemangioblastoma with retinal detachment and hemorrhage).45
Malformative Lesions
Malformative abnormalities are neoplastic and nonneoplastic masses that arise from an aberration of development.26,28,29,45,58 These are usually of neuroectodermal origin (e.g., dermoid or epidermoid) or mesodermal origin (e.g., lipoma). Benign teratomas can also be included in this category,59 as can vascular anomalies. Malformative lesions are often cystic but may be solid or of mixed consistency. In the orbital region in childhood, malformative lesions include colobomas, duplication cysts of the eye, nasolacrimal duct (NLD) cysts, lacrimal ectopia, dermoids, epidermoids, benign teratomas, and (rarely) arachnoid cysts and lipomas.
 FIGURE 3.15 Neurofibromatosis type I in a 17-year-old boy. Axial CT image shows left sphenoid wing dysplasia causing orbital deformity. There is also a meningoencephalocele extending through the deformed orbital apex into the orbit (black arrow), macrophthalmia (white arrow), and a plexiform neurofibroma overlying the lateral aspect of the left lid (arrowheads). |
Coloboma
Coloboma and colobomatous cyst have been discussed earlier. See section Malformations of the Globe.
Nasolacrimal Duct Cyst and Mucocele
Congenital NLD cyst and mucocele are the most common abnormalities of the infant lacrimal apparatus.60,61,62,63 They probably result from failure of canalization of the NLD. A residual membrane persists where the duct enters the nasal cavity, leading to partial or complete obstruction of the NLD, usually at the valve of Hasner, located beneath the inferior turbinate. Accumulation of secretions produces cystic NLD dilatation. Affected pediatric patients may present with a medial canthal mass due to a dacryocystocele, epiphora, nasal obstruction, and respiratory distress. It often resolves spontaneously in the early postnatal months; persistence may lead to nasal airway obstruction, infection, or dacryocystitis.
Imaging demonstrates a unilateral or bilateral cystic medial canthus mass in continuity with an enlarged NLD and an intranasal cystic mass (Fig. 3.16). Protrusion beneath the inferior turbinate distinguishes NLD cyst from cephalocele, which lies cephalad to the inferior turbinate.
 FIGURE 3.16 Nasolacrimal duct cyst and mucocele in a 3-month-old boy who was born with a “bluish mass” near the right lacrimal sac. CT images demonstrate a cystic mass (arrow) at the right medial canthus (A) that is contiguous with an enlarged nasolacrimal duct (B), with the mucocele extending into the inferior meatus of the nasal cavity (arrows in B). |
Lacrimal Gland Ectopia
Ectopic lacrimal gland tissue causes benign solid or cystic lesions of the orbit and may produce proptosis at any age.64 The ectopic tissue may be located intraconally.
Epibulbar Choristoma, Dermoid and Epidermoid Cysts, Teratoma
Choristoma is a broad term used to denote a congenital maldevelopmental lesion stemming from overgrowth of normal tissue in an abnormal location. The term choristoma can be applied to dermoid cyst, ectopic skin, ectopic bone, or other ectopic tissues. A collection of adipose tissue (lipoma) could be considered a choristoma or a hamartoma if it occurs in an anatomic site where adipose tissue is typically found. Epibulbar choristomas are variable in size, are unilateral or bilateral, and may be associated with the cornea, limbus, or subconjunctival space (Fig. 3.17).65 Epibulbar choristomas may be associated with ocular anomalies such as staphyloma, aniridia, congenital aphakia, and microphthalmia. They may coexist with choristomas of the eyelid, osseous choristomas of the choroid, orbital dermoids, choristomas of the face and scalp, and preauricular cartilage choristomas (i.e., accessory ear tissue). There is an association with the Goldenhar phenotype of hemifacial microsomia (HFM) and epidermal nevus syndrome.
 FIGURE 3.17 Epibulbar choristoma. Axial contrast-enhanced CT image shows a fat-density lesion (arrow) overlying the lateral corneal/conjunctival surface of the left globe. |
Dermoid cysts are the most common congenital lesions of the orbit.28,66,67,68 They arise from inclusion and sequestration of ectodermal elements, typically in sutures. Dermoids contain dermal adnexal structures such as sebaceous and sweat glands, hair follicles, and sometimes fat (Fig. 3.18), and squamous epithelium (Fig. 3.19). Epidermoid cysts have only keratinizing, stratified squamous epithelium and are rare in the pediatric orbit.69 In the orbit, dermoids typically occur laterally at the frontozygomatic suture and less commonly medially at the frontoethmoidal or frontonasal suture. Most present as a subcutaneous nodule near the orbital rim (Fig. 3.18B). These lesions demonstrate slow growth, with smooth bony remodeling and displacement of adjacent structures. On CT, they appear sharply defined and hypodense. On MR, they are of variable signal on T1-weighted sequences, are usually hyperintense on T2-weighted sequences, and typically have restricted diffusion and minimal if any peripheral enhancement. Notching of the bony site of origin and bony scalloping are characteristic.
Teratomas contain tissue derived from ectoderm, mesoderm, and endoderm. Orbital teratomas are rarely malignant but can cause significant proptosis in infancy.26,28,29,58 They are typically cystic and solid and can produce significant mass effect. Imaging reveals fat, soft tissue, calcification, and/or ossification.
Congenital Cystic Eye
A congenital cystic eye results when the optic vesicle does not invaginate normally during embryologic development.70 There is a large, cystic, often septated structure replacing the normal globe within an enlarged orbit. Incomplete rudimentary fragments of optic nerve and extraocular muscles may be present. On MRI, the fluid within the cystic septated structure exhibits a different signal than does normal vitreous, because it is filled with a more serous fluid.
 FIGURE 3.18 Orbital dermoid in a 9-year-old boy. A: Coronal CT image demonstrates a lobulated mass (arrow) that is predominantly of fat density but has some heterogeneous soft tissue in its inferior aspect. It is located in the superotemporal aspect of the orbit abutting the frontozygomatic suture and causes bony remodeling. The mass was surgically removed; pathology revealed a dermoid cyst with sebaceous glands. B: Coronal T2-weighted MR image in a different patient shows a well-circumscribed, lobulated cystic mass (arrow) overlying the superolateral orbital wall. This mass was also surgically excised and was pathologically confirmed to be a dermoid cyst. |
Infectious and Inflammatory Disorders
Preseptal and Postseptal Cellulitis; Orbital Subperiosteal Abscess
Orbital infection is common in children and usually results from acute complicated paranasal sinus infection. Orbital involvement occurs from direct extension via infected intervening bone or via emissary veins (Fig. 3.20), or because of complications of sinusitis such as a ruptured mucocele or mucopyocele (Fig. 3.21). Other sources include trauma, extension of facial or dental infection (Fig. 3.22), and, rarely, hematogenous spread from systemic infection.30 Infection is most often bacterial in origin, with Staphylococcus, Streptococcus, and Pneumococcus being the primary causative agents.71,72 Invasive fungal sinusitis can also spread to the orbit, especially in immunocompromised pediatric patients, primarily because of Mucor and Aspergillus. When infection affects only structures anterior to the orbital septum (skin, eyelids), it is termed preseptal (periorbital) cellulitis. When infection affects structures posterior to the septum, it is termed postseptal (orbital) cellulitis.
Pediatric patients with preseptal cellulitis typically present with rapid-onset soft tissue swelling and erythema, with or without pain, epiphora, and blurred vision. On imaging, there is diffuse soft tissue thickening of the skin and eyelids with adjacent fat stranding. The orbital septum tends to prevent postseptal spread of infection unless there is initiating sinus infection. The clinician should be alerted to sinus opacification, foreign object, or dental infection as a potential source of infection.
 FIGURE 3.19 Dermoid cyst in a 15-year-old girl. This dermoid cyst was resected from the floor of the mouth. The cyst was filled with yellow cheesy material, much of which remained stuck to the lining after sectioning. These contents are derived from the keratin debris and sebaceous secretions of the cyst wall, which show a squamous epithelial lining containing adnexal structures (not pictured). |
Postseptal cellulitis usually results from initiating bacterial sinus infection. Affected pediatric patients typically present with lid edema, proptosis, chemosis, and, if severe, impaired ocular motility. Postseptal extraconal cellulitis or phlegmonous change with or without subperiosteal abscess typically occurs in proximity to the infected sinus and thus occurs medially with ethmoid sinusitis (Fig. 3.20), inferiorly with maxillary antral sinusitis, and superiorly with frontal sinusitis. Contrast-enhanced CT demonstrates hazy increased density of the extraconal fat, with enlargement of extraocular muscles adjacent to extraconal cellulitis/phlegmon. Subperiosteal abscess appears as a low-attenuation, peripherally enhancing elliptical collection. Bone destruction, consistent with osteomyelitis, is sometimes seen. Spread of infection to the intraconal compartment results in orbital cellulitis with stranding of the intraconal fat. Dacryocystitis and dacryoadenitis can complicate or cause orbital infection.
 FIGURE 3.20 Orbital/periorbital cellulitis and subperiosteal abscess secondary to sinusitis. Axial contrast-enhanced CT image demonstrates ethmoid sinus opacification. There is a subperiosteal abscess (arrowhead) laterally displacing orbital contents and causing proptosis, and soft tissue thickening in the preseptal space (arrow). |
Invasive fungal sinusitis with spread to the orbits is seen predominantly in immunocompromised pediatric patients, with a high mortality of 50% to 80%.73 The responsible pathogens include Zygomycetes (e.g., Rhizomucor and Mucor) and Aspergillus species. The fungal organisms spread aggressively, with angioinvasion, bone invasion leading to bony destruction visible on CT, and hematogenous dissemination. Tuberculous infection is an important diagnostic consideration for destructive inflammatory sinonasal and orbital disease in endemic areas.
The most frequent vascular complication of orbital infection is thrombosis of the superior ophthalmic vein,9 which fails to enhance normally on contrast-enhanced CT or MR producing the “tram track” sign. Occasionally, severe postseptal cellulitis with significant infiltration and inflammation of the orbital fat and proptosis causes stretching and, rarely, vascular compromise of the optic nerve.
 FIGURE 3.21 Ruptured mucopyocele causing periorbital cellulitis. Axial contrast-enhanced CT image shows a frontal ethmoid mucocele (arrow) that has expanded into the periorbital and orbital region and ruptured, leading to spread of infection to the preseptal/periorbital space (arrowheads). |
 FIGURE 3.22 Odontogenic sinus infection leading to orbital/periorbital cellulitis. A: Axial CT image demonstrates extensive ethmoid and sphenoid sinus disease, which spread through the dehiscent lamina papyracea (arrow in A) into the orbit. B: Coronal CT image shows the source of the sinus infection. There is a periapical abscess around the root of a molar that has a dehiscent bony covering (arrow in B), rendering the abscess contents contiguous with the maxillary antrum. This is also the case on the left side, but there is milder left maxillary sinus disease. |
Complications of postseptal infection include osteomyelitis, epidural abscess, subdural empyema (Fig. 3.23), meningitis, cerebritis, parenchymal abscess, and cavernous sinus thrombophlebitis/thrombosis (Fig. 3.24). MRI is used to detect intracranial complications and osteomyelitis with T2-weighted fat-suppressed MR images, contrast-enhanced T1-weighted fat-suppressed MR images, DWI, and MRV. Rarely, steno-occlusive intracranial arterial complications occur, particularly in the presence of suppurative change and/or thrombophlebitis in the cavernous sinus.
Early treatment consists of appropriate antibiotics and, if necessary, surgical drainage of any abscess or obstructed sinus.
 FIGURE 3.23 Subdural empyema as a result of orbital cellulitis and sinusitis. A: Coronal CT image shows extensive pansinus inflammation and orbital cellulitis. Foci of gas in the anterior cranial fossa indicate spread of infection intracranially. A dense subdural empyema is seen (arrows in A). B: Coronal contrast-enhanced T1-weighted MR image demonstrates to greater advantage the subdural empyema with rim enhancement (white arrows in B) and orbital abscesses (black arrows). There is also a small subfrontal epidural abscess (asterisk). |
Idiopathic Orbital Inflammatory Syndrome
Idiopathic orbital inflammatory syndrome (IOIS), commonly referred to as orbital pseudotumor, is a nongranulomatous noninfectious benign inflammatory disease of the orbit with no identifiable cause, responsive to steroid therapy. IOIS is rare in children.74 Affected pediatric patients present with pain, swelling, erythema, proptosis, and/or diplopia. The classic finding is “painful ophthalmoplegia.” Vision may be impaired if there is optic nerve involvement. Iritis and papillitis are more commonly seen in children than in adults. IOIS is bilateral in up to one-third of pediatric cases without an identifiable underlying systemic condition.75 This is a diagnosis of exclusion, based on clinical history, disease course, and response to steroids. Histologically, there is an inflammatory/lymphoid infiltrate of orbital tissues acutely; in the subacute or chronic phase, fibroblasts and fibrosis are seen. However, biopsy is rarely performed as the diagnosis can usually be made clinically.
 FIGURE 3.24 Sinusitis, orbital cellulitis and abscess, and bilateral cavernous sinus thrombosis. Axial postcontrast T1-weighted fat-suppressed MR image shows ethmoid and sphenoid sinus inflammation and fluid, hazy enhancement in the orbital fat from orbital cellulitis, enhancement of the extraocular muscles compatible with myositis, and a focal orbital abscess (white arrow) on the right. In addition, there is expansion and nonenhancement of the cavernous sinus (small paired arrows), especially on the right, compatible with cavernous sinus thrombosis. There is narrowing of the internal carotid artery flow voids. Tentorial and dural enhancement is related to pachymeningitis (arrowheads), and enhancement of the temporalis muscles represents myositis (black arrows). This was a 14-year-old boy with a 3-week history of upper respiratory infection and headaches, diagnosed with sinusitis. This progressed to worsening fever, headache, and right eye swelling associated with vision loss. He eventually underwent ethmoidectomy and orbital decompression and was placed on antibiotic and anticoagulation therapy. |
IOIS may be classified according to the primary site of involvement: anterior or diffuse orbital inflammation, orbital myositis, perineuritis and periscleritis, lacrimal adenitis, and apical orbital inflammation (Fig. 3.25). The mass-like
manifestation of IOIS is also referred to as “inflammatory pseudotumor.” Painful external ophthalmoplegia, also known as Tolosa-Hunt syndrome, is considered a variant of IOIS that involves the orbital apex, superior orbital fissure, and cavernous sinus (Fig. 3.26). The radiographic differential diagnosis of Tolosa-Hunt syndrome includes fungal infection, sarcoidosis, lymphoma, and meningioma.
manifestation of IOIS is also referred to as “inflammatory pseudotumor.” Painful external ophthalmoplegia, also known as Tolosa-Hunt syndrome, is considered a variant of IOIS that involves the orbital apex, superior orbital fissure, and cavernous sinus (Fig. 3.26). The radiographic differential diagnosis of Tolosa-Hunt syndrome includes fungal infection, sarcoidosis, lymphoma, and meningioma.
 FIGURE 3.25 Idiopathic orbital inflammatory syndrome (IOIS) in a 17-year-old girl who presented with right orbital pain. Coronal CT image demonstrates infiltration of the orbital fat and enlargement of the inferior rectus muscle with hazy margins (arrow). |
On CT, depending on the site of involvement, IOIS may be characterized by preseptal soft tissue thickening and edema, orbital fat stranding, an enhancing orbital mass, enlargement of the extraocular muscles including their tendinous insertions, thickening of and stranding around the posterior globe or optic nerve sheath complex, enlargement and enhancement of the lacrimal gland, or soft tissue fullness in the orbital apex and cavernous sinus. MRI is helpful for evaluation of the cavernous sinus region for abnormal enhancement or mass lesions.76
Differential diagnosis is broad, because IOIS can affect any structure in the orbit and may be diffuse or mass-like. Orbital cellulitis, neoplasms including lymphoma and leukemia, sarcoidosis (Fig. 3.27), granulomatosis with polyangiitis (Wegener granulomatosis) (Fig. 3.28), and other granulomatous diseases may have similar imaging appearance. In a child, biopsy should be considered if there is an atypical clinical course, if the abnormality is refractory to steroid therapy, or if localized involvement raises the possibility of a malignancy such as lymphoma or rhabdomyosarcoma (RMS).74
After infection and other local and systemic causes have been excluded, systemic steroid therapy with a slow taper may be initiated and is considered first-line treatment.77 Recurrence of symptoms with steroid withdrawal is unusual in the pediatric population.78
 FIGURE 3.26 Tolosa-Hunt syndrome. Axial postcontrast T1-weighted fat-suppressed MR image shows thickening and enhancement in the left orbital apex and cavernous sinus (arrows). The patient presented with binocular diplopia. Physical examination revealed left third and sixth nerve palsies, Horner syndrome, and pain in the V1 and V2 distribution, indicating pathology involving the left cavernous sinus. |
 FIGURE 3.27 Orbital sarcoidosis. Coronal postcontrat T1-weighted MR image shows ill-defined enhancing soft-tissue in the superior left orbit, causing inferior displacement of the globe. The appearance is nonspecific. This area was biopsied, and pathology revealed noncaseating granulomas compatible with sarcoidosis. |
Other Inflammatory Disorders
Chorioretinitis is inflammation of the posterior uvea of the globe. It is usually caused by congenital (TORCH) infections, with cytomegalovirus and congenital toxocara being the most common pathogens in the neonatal age group. On imaging, the vitreous may appear hyperdense on CT and hyperintense on MRI, without a discrete mass.
 FIGURE 3.28 Orbital granulomatosis with polyangiitis (Wegener granulomatosis). Coronal CT image demonstrates soft tissue masses (arrows) in the left orbit that are nonspecific in appearance and could be inflammatory, infectious, or neoplastic in etiology. However, note destruction of the nasal septum (asterisk), which is compatible with granulomatosis with polyangiitis, that results from a vasculitic, necrotizing, and inflammatory process leading to necrosis of the septum and “saddle-nose deformity.” |
 FIGURE 3.29 Optic neuritis from multiple sclerosis (MS). A: Coronal T2-weighted fat-suppressed MR image demonstrates abnormally increased signal in the right optic nerve (arrow in A). Optic nerves should be isointense to white matter on all sequences, because they are essentially extensions of white matter. B: Coronal postcontrast T1-weighted MR image demonstrates abnormal enhancement of the right optic nerve (arrow in B). |
Ocular toxocariasis occurs as an intraocular inflammatory response to the death of the larva of the nematode Toxocara canis, sometimes leading to sclerosing endophthalmitis. It is usually unilateral and seen in older children.14 CT findings include homogeneous vitreal density corresponding to a detached retina, organized vitreous, and inflammatory subretinal exudate, similar in appearance to Coats disease and noncalcified retinoblastoma. On MRI, there is variable hyperintensity on T1- and T2-weighted images.
Optic neuritis is not uncommon in children. It is diagnosed using the same clinical criteria used in adults, including subacute vision loss, pain with eye movement, visual field deficits, and a relative afferent pupillary defect. In contrast to adults, children with optic neuritis more frequently have bilateral involvement, profound vision loss, and prominent disc swelling. Etiologies include viral, postviral, granulomatous, postradiation, posttraumatic, demyelinating (Fig. 3.29), neoplastic (e.g., leukemia), and unknown underlying processes.29 On imaging, the optic nerve appears hyperintense relative to normal white matter on T2-weighted MR images, is variably swollen, and enhances. In one series, the authors found that the risk of multiple sclerosis after a first episode of optic neuritis in children is increased if one or more white matter lesion is seen on a brain MRI performed at the time of initial presentation. None of the patients in their series with a negative brain MRI at presentation were diagnosed with MS for the duration that they were followed (88.5 months).79 The differential diagnosis includes neuromyelitis optica, in which optic neuritis, hypothalamic signal abnormality, and long-segment spinal cord involvement are MRI features.
Thyroid-Associated Orbitopathy
Graves disease is uncommon in children, and only a minority of pediatric patients have prominent ophthalmic manifestations,80 termed thyroid-associated orbitopathy (TAO), Graves ophthalmopathy, dysthyroid orbitopathy, or thyroid eye disease, where there is deposition of glycosaminoglycan and fibrosis of the extraocular muscles, and adipogenesis in the orbit.81
On imaging, one may see increase in orbital fat, enlargement of the extraocular muscles relatively sparing the tendinous insertions, hypodensity in the muscles (Fig. 3.30), and crowding of the optic nerve at the orbital apex. The inferior and medial rectus muscles are most commonly involved.30 Coronal images are best for assessing the extraocular muscles, orbital fat, and bony orbital walls for evidence of remodeling. Compressive optic neuropathy at the orbital apex can occur in pediatric patients with significant muscle enlargement. The differential diagnosis for extraocular muscle enlargement includes TAO, IOIS (muscle form), infectious myositis, dural arteriovenous fistula, carotid cavernous fistula, lymphoma/leukemia, metastasis, and acromegaly.30
Neoplastic Disorders
Retinoblastoma
Retinoblastoma (RB), a malignant tumor of the immature retina, is the most common primary intraocular tumor in children (80% of all primary ocular cancers), and the third most common intraocular tumor in all age groups. It occurs
in infancy, even in utero, with 95% of cases occurring before age 5 years.82 RB is rarely seen in older children.83 Tumors may show an endophytic growth pattern, with tumor growing toward the vitreous; it may have an exophytic growth pattern with tumor in the subretinal space, causing an overlying retinal detachment; or it may have a mixed growth pattern. A diffuse infiltrating, plaque-like form occurs less commonly, where the tumor is flat on the surface or beneath the retina, with no obvious mass or calcifications.83
in infancy, even in utero, with 95% of cases occurring before age 5 years.82 RB is rarely seen in older children.83 Tumors may show an endophytic growth pattern, with tumor growing toward the vitreous; it may have an exophytic growth pattern with tumor in the subretinal space, causing an overlying retinal detachment; or it may have a mixed growth pattern. A diffuse infiltrating, plaque-like form occurs less commonly, where the tumor is flat on the surface or beneath the retina, with no obvious mass or calcifications.83
 FIGURE 3.30 Thyroid-associated orbitopathy. Coronal CT image demonstrates enlargement of all the extraocular muscles and hypodensity in the muscles from glycosaminoglycan deposition. |
 FIGURE 3.31 Retinoblastoma. A: Axial CT image demonstrates a soft tissue mass (arrow) in the posterior right globe with calcification within it. B: Axial T2-weighted fat-suppressed MR image in a different patient reveals a large hypointense right intraocular soft tissue mass (arrow). |
RB develops as a result of a “two-hit” oncogenic mutation involving the Rb1 tumor suppressor gene, occurring between the 3rd month postconception and age 4 years, when retinoblasts are maturing. Both copies of the Rb1 gene have to be mutated for retinoblastoma to develop. In the familial form (40% of cases, autosomal dominant transmission), all germ cells have an existing mutation of one copy of the gene; tumor develops when the second copy mutates. In the sporadic form, the germ cells are normal, and the somatic retinal cell acquires mutations in both copies of the Rb1 gene.84 Affected pediatric patients with the familial form are more likely to have bilateral disease and multiple tumors. The average age of presentation is 7 months for bilateral cases and 24 months for unilateral cases.85 In pediatric patients with familial RB, second primaries include soft tissue sarcomas, osteosarcomas, carcinomas, CNS tumors, leukemias, uterine sarcomas, lung cancers, and skin cancers. In these patients, the second primary is a greater cause of death than the RB itself.86,87 Radiation exposure greatly increases the risk of development of a second primary, particularly if occurring before age 1 year.88 For this reason, CT is no longer recommended for imaging RB.
RB is a major diagnostic consideration in a child presenting with leukocoria, the most common presenting sign (60%), but it is a late sign, with high survival rate but low rate of globe salvage. Strabismus at presentation (20%) is considered an early sign with high survival rate and higher chance of globe salvage.
MRI is used for diagnosis, staging, and treatment monitoring.22 A combination of ophthalmoscopy, US, and MRI with gradient echo sequences has been shown to effectively detect calcifications. When intraocular calcification within a normal-sized or enlarged globe is present in a young child, RB must be suspected, as very few other simulating lesions contain calcification.89 The calcific foci are variable in size and number (Fig. 3.31A). The diffuse infiltrating plaque-like form, however, may have very little calcification. The tumor itself appears mildly to moderately hyperintense to vitreous on T1-weighted sequence and hypointense on T2-weighted sequence (Fig. 3.31B). Calcifications appear hypointense on MRI, especially on gradient echo T2-weighted and FSE T2-weighted sequences. There is generally moderate to significant tumoral enhancement.
Optic nerve invasion and orbital invasion increase the risk of metastatic disease.90,91 Massive choroidal invasion is considered the only other risk factor.92,93 With orbital extension, tumor can disseminate or extend intracranially. It is therefore important to include imaging of the entire orbit and the brain to evaluate for tumor spread.
Trilateral retinoblastoma refers to a primary midline intracranial tumor, usually in the pituitary or pineal region, in the presence of bilateral retinoblastoma (Fig. 3.32). When there are two midline tumors, the term quadrilateral retinoblastoma is sometimes used.82
 FIGURE 3.32 Trilateral retinoblastoma in a 2-year-old girl. Sagittal postcontrast T1-weighted MR image shows a large pituitary tumor in this child who has bilateral retinoblastoma (not shown). |
 FIGURE 3.33 Retinoblastoma. Retinoblastoma, forming a white-gray mass extending from the retina into the vitreous (left); microscopically, the tumor shows poorly differentiated cells with frequent mitotic and apoptotic figures indicating a rapid cell turnover (right; hematoxylin and eosin, original magnification, 600×). |
Grossly, retinoblastoma is characterized in its early stages by a white-gray nodule on the retina. Later, growth can extend through the retina causing detachment, and involve the vitreous humor. Microscopically, the tumor shows poorly differentiated cells with high nucleus-to-cytoplasm ratios, frequent mitotic figures, and frequent apoptotic cells (Fig. 3.33).
Treatment is best determined by a multidisciplinary team. Options include enucleation, cryoablation, laser photocoagulation, and chemothermotherapy.
Rhabdomyosarcoma
RMS is the most common primary nonocular orbital malignancy in children.29 RMS accounts for ˜50% of all pediatric soft tissue sarcomas and 15% of all pediatric solid tumors. Just over one-third of all RMSs are seen in the head and neck region, with the most common locations being the orbit (Fig. 3.34), masticator space, and paranasal sinuses (Fig. 3.35).26,29,45,58,68 For orbital RMS, the average age at diagnosis is 7 to 8 years, but it can be seen in older patients. In data collected by the Surveillance, Epidemiology, and End Results (SEER) program, the 5-year survival was found to be highest in children aged 1 to 4 years (77%) and worst in infants and adolescents (47% and 48%, respectively). Orbital site was the most favorable (86%).94 In children, the embryonal and alveolar subtypes predominate. Affected pediatric patients usually present with rapid-onset unilateral proptosis and globe displacement.
 FIGURE 3.34 Orbital rhabdomyosarcoma, embryonal subtype in a 5-year-old boy with proptosis. A: Coronal contrastenhanced CT image reveals a well-circumscribed enhancing mass (arrow) located within the superomedial right orbit with inferolateral displacement of the globe. The tumor is located primarily within the extraconal compartment. The superior rectus and superior oblique muscles are indistinguishable from tumor. B: Coronal T2-weighted fat-suppressed MR image shows that the tumor (arrow) is of lower signal than vitreous but relatively hyperintense compared with cerebral cortex. C: ADC map from axial DWI MR image demonstrates the tumor (arrow) showing decreased diffusion, attesting to its high grade nature. |
CT, MRI, and PET-CT are used to evaluate and stage RMS. Imaging of the head, chest, and abdomen is obtained for tumor staging. When the tumor is small, the margins are usually well defined; when large, the mass may have irregular margins and appear infiltrative, with surrounding bone and soft tissue invasion. On CT, it is usually isodense to muscle (Fig. 3.34). On MR, RMS is iso- or hypointense on T1-weighted sequence when compared to the brain.95 Tumors are of variable signal intensity compared to cerebral cortex and demonstrate decreased diffusivity. Internal hemorrhage is uncommon. The degree of enhancement is variable. CT is used to assess osseous involvement or destruction. MRI is superior for soft tissue delineation, for distinguishing tumor from sinus inflammation, for assessing bone marrow involvement, and for detecting intracranial extension or metastasis. Because there are no orbital lymphatics, nodal metastasis is rare except in very advanced cases.84
 FIGURE 3.35 Sinonasal rhabdomyosarcoma with orbital extension in a 17-year-old boy who presented with pansinusitis and proptosis. Coronal postcontrast T1-weighted fat-suppressed MR image shows a large infiltrative mass centered in the nasal cavity and extending into the orbits bilaterally, eroding orbital walls, and displacing and abutting orbital contents such as the extraocular muscles (arrows). There is also intracranial invasion (arrowheads). |
Differential diagnosis includes pseudotumor, lymphoma, leukemic infiltration, and metastatic disease. Neuroblastoma is a relatively common cause of orbital metastatic disease, typically in the first decade of life. Unlike RMS, there are usually multiple extraconal tumor masses with lytic permeative destruction of bone and spiculated periosteal reaction. Ultimately, tissue diagnosis is necessary to distinguish RMS from other noninflammatory solitary orbital tumors, with the exception of infantile hemangioma.
Treatment includes surgery, radiation, and/or chemotherapy. Whereas smaller tumors may be completely excised with a 5-year survival of ≥90%, larger more infiltrative tumors usually require postoperative radiation therapy to control residual disease. When there is significant residual tumor, 5-year survival is ≤35%. Combination chemotherapy helps to improve survival.
 FIGURE 3.36 Bilateral optic pathway glioma associated with neurofibromatosis type 1 (NF-OPG). Axial T1-weighted MR images (A and B) demonstrate fusiform enlargement of bilateral intraorbital, intracanalicular, and prechiasmatic optic nerves (arrows in A) with involvement of the optic chiasm and proximal optic tracts (arrows in B). |
Neurofibromatosis Type 1-Related Tumors
NF1 is an autosomal dominant disorder resulting from mutation of the neurofibromin gene on chromosome 17q11.2 and characterized by café au lait spots, axillary or inguinal freckles, neurofibromas and plexiform neurofibromas (PNFs), optic nerve gliomas, Lisch nodules in the eye, sphenoid bone dysplasia, and thinning of long bones.96 Orbital manifestations include orbital neoplasms (the most common of which is optic glioma [Fig. 3.11]), plexiform neurofibromas, orbital osseous dysplasia (Fig. 3.15), and congenital glaucoma.97
PNFs are transpatial and infiltrative. They arise from nerve fascicles, tend to grow along nerves, and may involve multiple nerve branches and plexuses, causing significant morbidity. There may be resultant thickening of periorbital soft tissues (Fig. 3.15), enlargement of the bony orbit, and extensive infiltration of the orbital soft tissues.98 Although they are more often extraconal,26 intraconal involvement produces increased density of the intraconal fat; enhancing, irregular nodular thickening of the optic nerve sheath complex; and thickening and enhancement of the uveal/scleral layer, believed to represent PNFs of these structures.98 PNFs have the potential to transform into malignant peripheral nerve sheath tumors (MPNSTs).
Optic pathway gliomas (OPGs) in NF1 are relatively benign and typically occur in children. Considered low-grade astrocytomas, they are distinct from the extremely rare malignant optic gliomas that are typically seen in adults.99 On imaging, an optic nerve glioma may be fusiform (Figs. 3.11 and 3.36) or exophytic; the nerve itself is enlarged and may be elongated, with kinking or buckling. On MR, the tumor is iso- to hypointense on T1-weighted sequence and iso- to hyperintense on T2-weighted sequence when compared to normal white matter (Figs. 3.11 and 3.36), with variable enhancement.100
Of the OPGs that are low-grade astrocytomas, different imaging features have been noted between those associated with NF1 (NF-OPG) and those not associated with NF1 (non-NF-OPG).101 In pediatric patients with NF-OPG, the most common site of involvement is the optic nerve, the original shape of the optic pathway is preserved, and cystic components are uncommon. In pediatric patients with
non-NF-OPG, the most common sites of involvement are the chiasm and hypothalamus, the tumor is larger and more masslike, and cystic components are more common. Pediatric patients with OPG may present with decreased vision or proptosis from mass effect. Additional symptomatology results from intracranial tumor. The differential diagnosis for mild enlargement of the optic nerve without enhancement includes the so-called “NF spot”, which is sometimes implicated if the lesion undergoes spontaneous regression.
non-NF-OPG, the most common sites of involvement are the chiasm and hypothalamus, the tumor is larger and more masslike, and cystic components are more common. Pediatric patients with OPG may present with decreased vision or proptosis from mass effect. Additional symptomatology results from intracranial tumor. The differential diagnosis for mild enlargement of the optic nerve without enhancement includes the so-called “NF spot”, which is sometimes implicated if the lesion undergoes spontaneous regression.
 FIGURE 3.37 Orbital lymphoma in a 9-year-old boy who presented with proptosis. A: Coronal T2-weighted fat-suppressed MR image shows a homogeneous, hypointense (relative to CSF), left superomedial extraconal mass (arrow) involving the left superior oblique muscle. B: Coronal postcontrast T1-weighted fat-suppressed MR image shows the homogeneously enhancing mass (arrow). |
The prognosis in children with NF-OPG is better than in those with non-NF-OPG, with the tumor often remaining quite stable in imaging appearance over a number of years. The current treatment includes surgery, chemotherapy, and radiation.
Orbital Lymphoma and Leukemia
There is a wide spectrum of orbital lymphoid lesions or lymphoproliferative disorders, ranging from reactive lymphoid hyperplasia to pseudotumor (IOIS) to malignant lymphoma. Reactive lymphoid hyperplasia and orbital lymphoma are essentially indistinguishable from each other on imaging75 and occur rarely in children. Malignant lymphoma may arise in the orbit, in the sinonasal region with extension into the orbit, or may be a systemic disease that includes an orbital focus. Any orbital structure may be involved, including the lacrimal gland, extraocular muscles, and orbital fat. On MR, lesions have intermediate to low signal on T1-weighted sequences and are isointense to cerebral cortex on T2-weighted sequences, with decreased diffusivity. The globe is rarely deformed by an adjacent lymphoma. Tumors occur in the anterior portion of the orbit, retrobulbar region, or superior orbital compartment (Fig. 3.37).
Leukemia is one of the most common pediatric malignancies and includes acute lymphoblastic leukemia, acute myelogenous leukemia (AML), and chronic myelogenous leukemia.102 Chronic lymphocytic leukemia is rare in children. Orbital involvement is mainly in the form of deposits of leukemic cells in the bone or soft tissue. These deposits are known as granulocytic sarcomas in the setting of AML, also called chloroma, because of the greenish color of myeloperoxidase on gross examination. Chloromas are usually seen in the subperiosteal region, in the lateral or medial orbital wall (Fig. 3.38). Dural or leptomeningeal disease may be present concurrently.102
Differential diagnosis of orbital lymphoma and a focal leukemic deposit includes RMS, Langerhans cell histiocytosis (LCH), and benign lymphoid disorders. The differential diagnosis for diffuse orbital leukemic involvement is metastatic disease, primarily neuroblastoma.
Langerhans Cell Histiocytosis
LCH is a clonal lesion of unknown etiology that is best classified as a neoplasm. LCH is characterized by proliferation and infiltration of abnormal histiocyte-like cells within various tissues. LCH preferentially affects children between the ages of 1 and 4, with disseminated or localized forms of disease that
have been variously referred to as Hand-Schüller-Christian disease, Letterer-Siwe disease, and eosinophilic granuloma.9
have been variously referred to as Hand-Schüller-Christian disease, Letterer-Siwe disease, and eosinophilic granuloma.9
 FIGURE 3.38 Chloroma in the orbit in a 17-year-old girl with a history of acute myelogenous leukema (AML) and chloromas at multiple locations in the body, including the spine and the abdomen. Axial CT image shows a homogeneous mass (arrow) along the lateral orbital wall. |
Lesions in children are mostly seen in the bone or bone marrow. Localized orbital involvement is not uncommon. Clinical signs and symptoms include proptosis, edema, erythema, and periorbital pain. Lesions are usually seen in the superior or superolateral orbital wall. Ocular involvement is rare.
On CT, LCH produces sharply defined lytic bone lesions with “punched-out” or beveled margins. On MR imaging, the associated soft tissue mass is of intermediate to low signal intensity relative to cerebral cortex on T2-weighted images with moderate to marked homogeneous or heterogeneous enhancement. There is often intracranial extension, involving the epidural space. Multiple lesions may be seen.9
The differential diagnosis for LCH includes ossifying fibroma, giant cell lesion, aneurysmal bone cyst (ABC), lymphoma, and metastasis. Ossifying fibroma is expansile with osseous matrix. Giant cell lesion and ABC are expansile lytic lesions, often with preservation of a thin rim of bone at the margin of the lesion on CT and characteristic fluid-fluid levels on MR.
Vascular Lesions
Orbital vascular lesions include hemangiomas (infantile, congenital, and other), high-flow malformations (arteriovenous malformations [AVMs] and cavernous carotid fistulas [CCFs]), low-flow malformations (venous malformations, lymphatic malformations, and combined malformations), and varices. Only the hemangiomas are considered true neoplasms.103 The term “cavernous hemangiomas” is a commonly used misnomer for well-encapsulated low-flow venous malformations as determined histopathologically. Vascular malformations and tumors are discussed in detail in other sections of this book. The imaging features of orbital vascular anomalies are briefly discussed and illustrated below.
 FIGURE 3.39 Infantile hemangioma in two different pediatric patients. A: Axial T2-weighted MR image demonstrates a mass in the medial right orbit causing mass effect, displacing the globe laterally and causing proptosis. There are prominent internal flow-voids (arrows), which, along with avid homogeneous enhancement (not shown), are diagnostic of a hemangioma. This 2-month-old boy presented with right upper lid swelling that began at age 2 weeks, with the lesion rapidly enlarging over the following weeks. B: Axial postcontrast T1-weighted fat-suppressed MR image shows an avidly enhancing preseptal and intraorbital hemangioma (short arrows), a large right temporalis muscle hemangioma, and a hemangioma within the right internal auditory canal (arrowhead). Note the ipsilateral cerebellar hypoplasia (long arrow) in this patient with PHACES association. This 1-monthold boy presented with multiple hemangiomas. |
Infantile Hemangioma
The imaging appearance of infantile hemangiomas depends on the clinical stage. During the proliferative phase, a lobulated, sharply defined, homogeneous, highly vascular soft tissue mass is seen, with intense enhancement. On MRI, proliferating hemangioma is iso- or hypointense to muscle on T1-weighted MR images, moderately hyperintense with vascular flow voids on T2-weighted MR images (Fig. 3.39), and intensely enhancing.104 During the involuting phase, enhancement and vascularity diminish. After complete involution, a focal region of fibrofatty tissue remains.
A subgroup of patients with infantile hemangiomas has structural anomalies elsewhere in the body, with involvement of the brain, cerebral vasculature, aorta, eyes, and chest wall. In 1978, Pascual-Castroviejo et al., described the association of facial and scalp “hemangiomas” with brain abnormalities, malformations of the extra- and intracranial blood vessels, and congenital heart disease.105 Subsequently, Frieden et al. coined the acronym “PHACES”106 denoting the major features of this neurocutaneous association: posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, eye abnormalities, and sternal clefting and supraumbilical abdominal raphe. The most common posterior fossa malformation is unilateral cerebellar hypoplasia with a prominent ipsilateral CSF space. Intracranial arterial anomalies include persistent fetal anastomotic connections (e.g., persistent trigeminal artery), aplasia or hypoplasia of the carotid or vertebral arteries, and dilatation and tortuosity of the carotid or cerebral arteries. Steno-occlusive changes with moyamoya collaterals have also been observed.107 Eye findings include microphthalmia, optic nerve hypoplasia, congenital cataracts, morning glory anomaly, and increased retinal vascularity. The unilateral or bilateral hemangiomas in patients with PHACES tend to be large, regional, and plaque-like, sometimes in a beard-like distribution or in the midline. Therefore,
pediatric patients with large infantile hemangiomas in the face or head and neck region should undergo brain MRI and MRA to evaluate for PHACES association.108
pediatric patients with large infantile hemangiomas in the face or head and neck region should undergo brain MRI and MRA to evaluate for PHACES association.108
 FIGURE 3.40 Lymphatic malformation. A: Axial postcontrast T1-weighted fat-suppressed MR image demonstrates a heterogeneous lesion (arrows) in the retrobulbar orbit causing proptosis. There are areas of patchy enhancement, corresponding to venous components. B: Axial T2-weighted MR image more rostrally shows fluid-fluid levels (arrows), characteristic of lymphatic malformations. |
Vascular Malformations
High-flow vascular malformations include AVMs and arteriovenous fistulae (AVF). Low-flow vascular malformations include venous malformations (VMs), lymphatic malformations (LMs), and combined malformations.
VMs are well-defined, transpatial multiloculated cystic masses. The venous lakes typically appear hyperintense on T2-weighted MR images, similar in signal to CSF, with gradual enhancement of the contained venous blood with contrast. The presence of rounded signal voids on MR or calcific foci on CT due to phleboliths is a diagnostic feature of VMs.
Orbital LMs present with proptosis that is sometimes apparent at birth or that manifests subsequently because of rapid enlargement from intercurrent infection or hemorrhage. LMs are typically transpatial lesions. LMs with cysts larger than 1 cm in size are termed macrocystic and are sharply defined. Microcystic LMs consist of small or tiny cysts that are less well defined and sometimes appear more infiltrative. Components of the lesion insinuate within and between normal tissues and structures of the lid and orbit. Cyst contents are hyperintense on T2-weighted MR images unless complicated by hemorrhage, in which case hypointense blood products will be seen. Signal intensity on T1-weighted MR images varies depending on protein content and chronicity of hemorrhage. Macrocystic LMs appear as distinct large locules of fluid spaces separated by enhancing septations. Microcystic LMs may appear to be enhancing without appreciable fluid-filled cystic spaces. Fluid-fluid levels within the cystic spaces related to blood products of various ages are characteristic of LMs (Fig. 3.40).
 FIGURE 3.41 Venous malformation (VM) in a 20-year-old woman with multiple venous malformations and a diagnosis of blue rubber bleb nevus syndrome. A: Axial CT image shows a phlebolith (arrow) in the lobulated left orbital mass. B: Axial FSEIR image shows hyperintense VMs within the intraconal compartment of the left orbit and within the temporalis muscle and scalp (short arrows). Note the associated intracranial developmental venous anomaly (long arrow). C: The VMs (arrows) enhance homogeneously only on delayed postcontrast T1-weighted fat-suppressed MR image acquired several minutes after the administration of contrast. |
Many of the orbital low-flow vascular anomalies demonstrate features of both LM and VM and are thus considered combined lesions. Interestingly, these lesions are associated with a significant incidence of intracranial venous anomalies (cavernous malformations and prominent developmental venous anomalies), (Fig. 3.41) and even occasionally AVF.109
Orbital AVM and AVF are uncommon high-flow lesions that produce pulsatile proptosis and sometimes a bruit. On imaging, they are characterized by enlarged arterial feeders
and early draining veins. AVM is distinguished from AVF by the presence of a tangle of small vessels known as a nidus between the feeding and draining vessels.
and early draining veins. AVM is distinguished from AVF by the presence of a tangle of small vessels known as a nidus between the feeding and draining vessels.
 FIGURE 3.42 Orbital varix. Coronal contrast-enhanced CT image demonstrates a dilated vascular space that fills with contrast (arrow). On axial CT images, this structure can be seen to be contiguous with a vein proximal and distal to it (not shown). |
Orbital Varix
A primary orbital varix presumably occurs as a result of congenital weakness of the venous wall, resulting in dilatation of one or more orbital veins. A tangled mass of venous channels overlaps with the spectrum of VM. With Valsalva maneuver, the varix enlarges and may lead to increased proptosis or globe displacement.84 Orbital varices may be associated with intracranial vascular anomalies or malformations, including arteriovenous shunts. On CT, calcifications representing phleboliths may be seen. On MR, they are hyperintense on T2-weighted MR images and enhance. Unless thrombosed, they should fill in with contrast at the venous phase (Fig. 3.42).
Cavernous Carotid Fistula
CCF occurs when there is a tear in the cavernous portion of the internal carotid artery, allowing arterial blood to enter the cavernous sinus, leading to increased cavernous sinus pressure and reversal of blood flow in the veins that normally drain into the cavernous sinus. Affected pediatric patients with CCFs present with proptosis, chemosis, pulsating exophthalmos, an auscultable bruit, and/or objective pulsatile tinnitus. On imaging, there is engorgement of the superior ophthalmic vein (Fig. 3.43) and enlargement of the ipsilateral extraocular muscles. Venous thrombosis of the cavernous sinus or superior ophthalmic vein may occur. Mimics of CCF include AVMs and AVFs; isolated dilatation of the superior ophthalmic vein can also occur.110 Therefore, correlation with clinical history and examination is paramount. Diagnosis of CCF can be confirmed with CTA, MRA, or conventional catheter angiography.
 FIGURE 3.43 Cavernous carotid fistula. Axial contrast-enhanced CT images demonstrate engorgement of the left cavernous sinus (arrow in A) and left superior ophthalmic vein (arrow in B). |
Traumatic Disorders
Orbital injuries can occur from direct blunt or penetrating trauma. Common injuries include traumatic hyphema, globe rupture, vitreous hemorrhage, lens rupture or dislocation, ocular detachments, intraorbital foreign bodies, CCFs, optic nerve injuries, and osseous fractures. Terson syndrome is a vitreous or retinal hemorrhage occurring secondary to a subarachnoid hemorrhage, thought to result from a sudden large increase in intracranial pressure.111 Retinal hemorrhages are also seen as an important manifestation of inflicted head trauma.
CT is the modality of choice for imaging orbital trauma, demonstrating fractures, hemorrhage, and soft tissue injuries. Radiopaque (e.g., metal) or radiolucent (e.g., wood) foreign objects are also well visualized by CT, whereas MRI is contraindicated if metallic foreign object is suspected. There should be a conscious effort to minimize radiation exposure in children, particularly to the lens, while optimizing the protocol to allow the radiologist to make an accurate diagnosis. CT protocol should consist of thin-section axial scans with multiplanar reformations. US may be useful for evaluating the globe and its contents, but it is contraindicated if there is a possibility of a ruptured globe.110
Anterior Chamber Injuries
Traumatic hyphema (posttraumatic bleeding into the anterior compartment) results from disruption of the iris or ciliary body vessels, leading to extravasation of blood into the anterior compartment. On CT, the anterior compartment appears hyperdense. Corneal laceration leads to decreased
anterior compartment volume, manifested as decreased anteroposterior dimension of the anterior compartment.
anterior compartment volume, manifested as decreased anteroposterior dimension of the anterior compartment.
 FIGURE 3.44 Ruptured globe in two different pediatric patients. A: This patient was a 13-year-old boy who was hit in the eye by glass shards. Axial CT image shows contour deformity to the right globe. The globe is shrunken, with a flattened posterior margin (arrow). The lens (arrowhead) is also ruptured, appearing foreshortened with loss of the normal biconvex shape. B: This 5-year-old girl was sprayed in the eye with a pressure sprayer. Physical examination showed corneal laceration, hyphema, and subconjunctival hemorrhage. Axial CT image demonstrates, intraocular gas (arrow) and vitreous hemorrhage (arrowhead). The lens is also dislocated, no longer seen in its normal anatomic location. |
Globe Injuries
Globe rupture, also termed open globe injury, is an ophthalmologic emergency. It is a major cause of blindness, and prompt diagnosis and treatment are crucial to prevent further injury. Unrecognized globe rupture may rarely result in bilateral blindness due to sympathetic ophthalmia, a bilateral diffuse granulomatous intraocular inflammation that occurs following unilateral ocular surgery or penetrating trauma.
The injured eye is known as the exciting eye, and the contralateral eye is known as the sympathizing eye.112 The time from ocular injury to onset of sympathetic ophthalmia ranges from a few days to decades, with 80% occurring within 3 months and 90% within 1 year.113,114 It is believed to occur as a result of autoimmune inflammatory response to ocular antigens that become exposed to the immune system following loss of globe integrity. Therefore, if the injured globe is nonviable, or if there is little chance of the injured globe regaining visual function following trauma, prompt enucleation may help prevent the occurrence of sympathetic ophthalmia.
On CT, a ruptured globe is suggested by abnormality in the globe contour (Fig. 3.44A), loss of normal globe volume, scleral discontinuity, intraocular gas (Fig. 3.44B), or intraocular foreign objects.110 Decreased anterior chamber depth suggests an anterior globe laceration. With increased anterior chamber depth, traumatic rupture of the posterior sclera should be suspected. A discontinuity in the posterior sclera allows vitreous to prolapse through the defect, leading to decompression of the vitreous and posterior sagging of the lens.115 With vitreous hemorrhage, hazy diffuse or mass-like hyperdensity may be seen in the posterior chamber.
Lens Injuries
When there is traumatic deformity of the globe, the zonule fibers of the lens can stretch and tear, resulting in dislocation of the lens. Posterior dislocation is more common than anterior dislocation, in part because the iris prevents the lens from migrating anteriorly. If only some of the fibers are torn, then the lens may remain in position on one side, while angling posteriorly on the opposite side where it projects into the vitreous humor (Fig. 3.45).110
With blunt trauma, there may be tearing or rupture of the lens capsule. When that occurs, fluid enters the crystalline lens, normally the least hydrated organ of the body. On CT, therefore, a ruptured lens may retain a relatively normal contour but appear less dense than usual (Fig. 3.46).
Ocular Detachments
There are three main types of detachments in the globe: posterior hyaloid detachment (posterior vitreous detachment), retinal detachment, and choroidal detachment.
In posterior hyaloid detachment, the vitreous body becomes separated from the retina, and vitreous can dissect into the potential space between the posterior hyaloid membrane and the retina.84 Traction on the retina at points that are still attached increases the risk of subsequent retinal tears and detachment. The detached posterior hyaloid membrane
may be thickened and become visible on imaging. This condition may be seen in children with PHPV.
may be thickened and become visible on imaging. This condition may be seen in children with PHPV.
 FIGURE 3.45 Lens dislocation in a 13-year-old boy who was hit in the eye by a paint ball. Axial CT image shows the lateral portion of the lens dislocated (arrow) and angled posteriorly, whereas the medial portion remains anchored. |
 FIGURE 3.46 Lens rupture in a 9-year-old girl. Axial CT image demonstrates normal contour of the lenses bilaterally. However, the left lens (arrow) is less dense than normal, indicating the presence of fluid that has infiltrated into the lens through a torn capsule. |
Retinal detachment is separation of the sensory retina from the retinal pigment epithelium. Serous or exudative detachment usually occurs as a result of breakdown of the blood-retina barrier due to tumor, inflammation, vasculopathy (e.g., Coats disease), hematologic disorders, or in association with congenital anomalies such as optic nerve colobomas and morning glory syndrome.116 When there is scarring or another process in the vitreous causing traction of the sensory retina from the pigmented layer, it is referred to as a traction detachment. With trauma, there may be subretinal hemorrhage, with or without a tear in the sensory retina. The presence of retinal hemorrhage in a child should raise suspicion for nonaccidental trauma.110
The retina is a very thin structure and is not visible per se on imaging. Retinal detachment is diagnosed on imaging based on the difference in appearance of the normal vitreous and fluid accumulated in the subretinal space. The subretinal fluid typically appears homogeneously hyperdense, with a lentiform or V shape, extending from the ciliary body anteriorly to the optic disc posteriorly (Fig. 3.47). If the subretinal fluid happens to be isodense/isointense to vitreous, it may escape imaging detection. US is considered a better modality for imaging retinal detachment than CT or MR.117
Choroidal detachment is separation of the choroid from the sclera. Serous or hemorrhagic fluid can then accumulate in the suprachoroidal space. The latter is seen in the setting of trauma; the former occurs in the setting of ocular hypotony, which may result from inflammation, traumatic perforation, or surgery.110 Suprachoroidal fluid collections appear biconvex, extending from the level of the vortex veins to the ora serrata (Fig. 3.48). Their CT and MR appearance depends on their composition and may be variably dense/intense. Choroid is thick, and the separated choroid is often visible on CT and MR.
 FIGURE 3.47 Retinal detachment. Axial CT image demonstrates hyperdense fluid (arrows) with a wide V shape in the posterior right globe. |
Intraorbital Foreign Objects
MR is absolutely contraindicated if there is any suspicion of a potential intraorbital metallic object, as blindness may result if the patient is subjected to MR scanning because of migration of the object within the magnetic field.
Optic Nerve Injury
The optic nerve can be directly injured in penetrating trauma, by bone fragments causing nerve laceration, or from a nerve sheath hematoma. It can also be indirectly injured from transmission of
forces to the optic nerve following blunt trauma or from vascular compromise. For evaluation of the optic nerve, MRI is necessary, after confirming the absence of any potential intraorbital metallic objects. Normally, the optic nerve should follow white matter signal. If there is disruption of the normal nerve contour, or if there is abnormally hyperintense signal in the optic nerve on T2-weighted sequence, optic nerve injury should be suspected.
forces to the optic nerve following blunt trauma or from vascular compromise. For evaluation of the optic nerve, MRI is necessary, after confirming the absence of any potential intraorbital metallic objects. Normally, the optic nerve should follow white matter signal. If there is disruption of the normal nerve contour, or if there is abnormally hyperintense signal in the optic nerve on T2-weighted sequence, optic nerve injury should be suspected.
 FIGURE 3.48 Choroidal detachment. Axial CT image demonstrates biconvex dense fluid collections (arrows) on the medial and lateral surfaces of the right globe, not extending to the optic disc. |
 FIGURE 3.49 Intraocular metallic fragment. Axial CT image clearly shows a punctate metallic fragment (arrow) in the right globe. |
FACE, NASAL CAVITY, PARANASAL SINUSES
Embryology and Development
Facial structures develop from several primordia that surround the stomodeum (primitive mouth) at 4 to 5 weeks’ gestational age.119 Between 4 and 8 weeks’ gestational age, the nasomedial and maxillary processes fuse to form the upper lip and jaw. The nasomedial processes fuse and give rise to the philtrum of the lip, the premaxillary component of the upper jaw, and the primary palate. The frontonasal prominence gives rise to the forehead, nose, and nasal septum. The maxillary processes form parts of the upper lip, maxilla, and the secondary palate. Between the maxillary process and the nasal primordium is the nasolacrimal groove, which eventually gives rise to the NLD and the lacrimal sac. The bilateral mandibular prominences give rise to the mandible, lower lip, chin, and lower cheek. As basic facial structures are established, mesodermal cells associated with the first and second pharyngeal arches give rise to the muscles of mastication (first arch derivatives, innervated by cranial nerve V) and the muscles of facial expression (second arch derivatives, innervated by cranial nerve VII).
The nasal cavities communicate with the nasopharynx and oral cavity after rupture of the oronasal membrane at the choanae. Turbinates form from the lateral walls of the nasal cavities. Paranasal sinuses form as diverticula of the walls of the nasal cavities and undergo pneumatization after birth.
Normal Anatomy
The nose and paranasal sinuses continue to develop through puberty. The vestibule is the opening to each nasal cavity. The nasal septum consists of cartilage anteroinferiorly and bone (vomer and perpendicular plate of the ethmoid) posterosuperiorly. The superior, middle, and inferior turbinates arise from each lateral nasal wall. Supreme turbinates are variably present in some individuals.
The maxillary and ethmoid sinuses are pneumatized at birth and continue to develop until age 10 to 12 years. The sphenoid sinuses are pneumatized at age 1 to 2 and reach adult size by age 14 years. The frontal sinuses are difficult to distinguish from anterior ethmoid air cells until they become larger and extend above the superior orbital rims at age 8 to 10 years. In some individuals, the frontal sinuses never enlarge and remain nonpneumatized or underpneumatized.
The frontal, anterior and middle ethmoid, and maxillary sinuses drain into the middle meatus via the ostiomeatal complex (OMC). The posterior ethmoid and sphenoid sinuses drain into the sphenoethmoidal recess and superior meatus of the nasal cavity.
Not infrequently, ethmoid air cells expand beyond the confines of the ethmoid bone and encroach into the frontal, maxillary, and sphenoid regions, as well as deep to the lacrimal bone and into the frontal process of the maxilla. These anatomic variants include (1) agger nasi cells—anterior ethmoid air cells pneumatizing the frontal process of the maxilla; (2) supraorbital ethmoid cells—anterior ethmoid air cells pneumatizing the roof of the orbit; (3) Haller cells—infraorbital ethmoid air cells; (4) Onodi cells—posterior ethmoid air cells extending along the sphenoid bone, located superior to the sphenoid sinus and abutting the optic canal; and (5) a posterior ethmoid air cell extending along the maxillary sinus and separated from the maxillary sinus by a bony wall, causing a “double antrum.” Variant anatomy must be recognized in order to plan for surgical drainage targeting these particular cells should they be inflamed.
Congenital and Developmental Anomalies
Nasal Obstruction—Complete Nasal Stenosis, Choanal Atresia, Pyriform Aperture Stenosis
Neonates are obligate nose breathers, and obstruction of the nasal passages leads to airway compromise.120 Congenital causes include pyriform aperture stenosis, nasal cavity stenosis, and choanal stenosis or atresia. Large NLD cysts, craniofacial anomalies, and cephaloceles also cause obstruction. Stenosis of the entire nasal airway is usually bony and may be seen with maxillary hypoplasia.
Choanal atresia is congenital obstruction of the posterior choanae. Incidence has been reported to be between 1 in 5,000 and 1 in 9,000 live births. It is seen more often in females and is more commonly unilateral. Obstruction is invariably bony, with or without a membranous component. Recent literature suggests that mixed bony/membranous choanal atresia occurs in 70% of cases and pure bony obstruction in 30% of cases.121
There is also medialization of the pterygoid plates and lateral nasal walls (Fig. 3.50).
There is also medialization of the pterygoid plates and lateral nasal walls (Fig. 3.50).
 FIGURE 3.50 Choanal atresia in an 11-year-old boy who presented with chronic sinusitis and obstruction. Axial bone window CT image demonstrates narrowing of the right posterior choana (arrow), with a bony obstruction. The vomer is lateralized (as shown on figure), and the pterygoid plates are medialized (not shown). |
There are sometimes associated skull base abnormalities or defects.122 Approximately half of the affected pediatric patients have associated craniofacial anomalies such as CHARGE syndrome. CHARGE is an acronym for coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, and ear anomalies/deafness. Major criteria (ocular coloboma; choanal atresia; characteristic ear abnormalities— particularly absent or severely hypoplastic semicircular canals (SCCs), hypoplasia of the vestibule, ossicular and other middle ear abnormalities (Fig. 3.51)123; cranial nerve hypoplasia/aplasia) and minor criteria (cardiovascular malformations; genital hypoplasia; cleft lip/palate; tracheoesophageal fistula; hypothalamo-hypophyseal dysfunction; distinctive CHARGE facies; developmental delay) have been described. Individuals with all four major characteristics or three out of four major and three out of seven minor characteristics are reported to be highly likely to have CHARGE syndrome.124
 FIGURE 3.51 Semicircular canal hypoplasia/aplasia in CHARGE association. A: Axial bone window CT image shows a hypoplastic vestibule with only a rudimentary posterior semicircular canal (SCC) (short arrow). The lateral and superior SCCs are absent. There is only a single cochlear turn (long arrow), and the cochlear nerve canal is absent, resulting in an “isolated cochlea.” B: Coronal bone window CT image (different patient) through the middle ear demonstrates oval window stenosis. The tympanic segment of the facial nerve is abnormally inferiorly placed, overlying the stenotic oval window (white arrow). The superior semicircular canal is dysplastic, appearing as a bulbous cystic space (black arrow). The posterior and lateral semicircular canals were absent and the cochlea formed only 1.5 turns (not pictured). |
CT should be performed with the axial plane parallel to the hard palate at the level of the pterygoid plates. CT demonstrates obstruction of the posterior nasal cavity, characterized by medial bowing and thickening of the lateral nasal walls, enlargement of the posterior vomer, and a bony or membranous structure obstructing the choana representing the atretic plate. Bilateral choanal atresia should prompt inspection of the temporal bones for the typical diagnostic inner ear findings of CHARGE syndrome.
Syndromic craniosynostosis, TCS, and other craniofacial syndromes are associated with midnasal cavity stenosis due to midfacial hypoplasia.
Pyriform aperture stenosis is narrowing at the level of the anterior nasal cavity. On CT, there is marked stenosis of the pyriform apertures associated with a triangular morphology of the anterior hard palate. Images should include the maxillary dentition. If there is a single central megaincisor, then there is an association with holoprosencephaly and MR of the brain should be obtained (Fig. 3.52).
Lacrimal Sac Mucocele and Nasolacrimal Duct Mucocele
Lacrimal sac and NLD mucocele is described earlier in the Orbit section. NLD mucocele is common, especially in premature infants, and is an important cause of neonatal nasal obstruction. Most cases resolve spontaneously. Less commonly, pressure buildup may cause the imperforate distal membrane to balloon out into the nasal cavity, leading to an intranasal cystic mass and nasal obstruction. The entire enlarged cystic structure extending from the lacrimal sac to the inferior meatus is referred to as a nasolacrimal mucocele. This is well delineated on CT and MR, which demonstrate a dilated cystic structure at the medial canthal region contiguous with an expanded NLD, terminating in an intranasal submucosal cystic mass that projects beneath the inferior turbinate (Fig. 3.16).
 FIGURE 3.52 Pyriform aperture stenosis associated with holoprosencephaly. Axial bone window CT images (A and B) show narrowing of the pyriform aperture bilaterally (black arrows in A) and a single central megaincisor (white arrow in B). The patient went on to have an MRI. Axial T2-weighted MR image (C) shows absence of the anterior falx and interhemispheric fissure, fusion of the frontal lobes, and partial fusion of the thalami, compatible with semilobar holoprosencephaly. |
Glabellar/Nasal Pit, Dermal Sinus, Dermoid Cyst, Meningoencephalocele, and Nasal Neuroglial Heterotopia
During fetal development, the frontal bone is separated from the nasal bones by a fontanelle called the fonticulus frontonasalis. The nasal bones are in turn separated from the underlying cartilaginous nasal capsule by the prenasal space. The prenasal space extends up to the base of the brain and down to the nasal tip. Normally, projections of dura around the brain extend through these spaces in the midline and contact the ectoderm, then regress back toward the brain before these spaces are obliterated. By birth, the frontonasal suture is in the location of the obliterated fonticulus frontonasalis, the nasal cartilages and ethmoid bone are in the region of the obliterated prenasal space, and the frontal and ethmoid bones come together around a small ostium at the skull base called the foramen cecum, just anterior to the crista galli. If the embryonic dural projections do not regress properly, anomalies result.
Glabellar/Nasal Pit, Dermal Sinus, and Dermoid Cyst
As the dura regresses, it can bring with it adherent ectoderm, leading to the formation of a dermal tract. The tract can run either (1) from the skin surface of the glabella through the fonticulus frontonasalis (frontonasal suture) toward the foramen cecum or (2) from the skin surface of the nose through the prenasal space (under or through the nasal bones) toward the foramen cecum. Clinical manifestations include cutaneous pits, dermal sinuses, (epi)dermoid cysts, and fibrous cords. Rarely, these sinus tracts, cysts, and cords may extend intracranially and be adherent to the brain. With persistent intracranial communication, recurrent meningitis, abscess, or empyema may result.
Imaging should include the entirety of the potential course of a sinus tract based on embryologic anatomy—the skin surface, nose, and nasal cavity, to the anterior cranial fossa and foramen cecum (Fig. 3.53). The examination of choice is MR, and pulse sequences should include multiplanar thinsection, small field of view, high-resolution T2-weighted
fat-suppressed MR images, T1-weighted fat-suppressed MR images, postcontrast T1-weighted fat-suppressed MR images, and DWI. Thin-section axial CT images with multiplanar reformats should be obtained if bony anatomy evaluation is required. CT in infants under 2 years of age should be obtained with contrast in order to demonstrate the enhancing cartilage of the incompletely ossified anterior skull base that could otherwise be mistaken for a bony defect. On CT, the dermoid cyst and/or sinus tract appears as a rounded or tubular hypodense structure. A fibrous channel is difficult to detect. Rim enhancement may occur, but surrounding edema suggests superimposed infection.125 If a cyst or sinus extends close to or through the foramen cecum, the foramen may appear enlarged, and the crista galli may appear thickened, enlarged, and grooved or bifid.126 On MR, dermoids appear hypo-, iso-, or hyperintense on T1-weighted MR images, and tend to be hyperintense on T2-weighted MR images with decreased diffusivity. Occasional lesions demonstrate fat suppression. Dermal sinus tracts are usually of similar signal to CSF on all pulse sequences.
fat-suppressed MR images, T1-weighted fat-suppressed MR images, postcontrast T1-weighted fat-suppressed MR images, and DWI. Thin-section axial CT images with multiplanar reformats should be obtained if bony anatomy evaluation is required. CT in infants under 2 years of age should be obtained with contrast in order to demonstrate the enhancing cartilage of the incompletely ossified anterior skull base that could otherwise be mistaken for a bony defect. On CT, the dermoid cyst and/or sinus tract appears as a rounded or tubular hypodense structure. A fibrous channel is difficult to detect. Rim enhancement may occur, but surrounding edema suggests superimposed infection.125 If a cyst or sinus extends close to or through the foramen cecum, the foramen may appear enlarged, and the crista galli may appear thickened, enlarged, and grooved or bifid.126 On MR, dermoids appear hypo-, iso-, or hyperintense on T1-weighted MR images, and tend to be hyperintense on T2-weighted MR images with decreased diffusivity. Occasional lesions demonstrate fat suppression. Dermal sinus tracts are usually of similar signal to CSF on all pulse sequences.
 FIGURE 3.53 Dermoid and dermal sinus (median nasal dermal fistula) in a 9-month-old boy who presented with a small mass overlying the nose. Sagittal T2-weighted MR image shows a rounded, hyperintense dermoid abutting the nasal bone (long white arrow) connected by a tubular dermal sinus tract (short white arrow) to a dermoid located at the foramen cecum (black arrow) projecting into the epidural space. There is also a dermoid in the nasal septum (arrowhead). |
Meningoencephalocele and Nasal Neuroglial Heterotopia
The dural projection may fail to regress, resulting in a patent diverticulum that communicates with the intracranial compartment. The diverticulum may contain leptomeninges, CSF, and/or neural tissue, resulting in a meningocele or meningoencephalocele, broadly referred to as cephalocele. A nasofrontal cephalocele protrudes through a persistent fonticulus; a nasoethmoidal cephalocele protrudes through a persistent prenasal space. If the diverticulum becomes closed off and loses its communication with the intracranial compartment, it forms an isolated mass of heterotopic meninges and neural tissue, termed nasal neuroglial heterotopia (“nasal glioma”). Extranasal neuroglial heterotopia comes from tissue that protruded through the fonticulus frontonasalis. Intranasal neuroglial heterotopia comes from tissue that protruded through the prenasal space.
Imaging is helpful for evaluating the location and size of the mass and its relationship with the brain and foramen cecum. On MRI, it is usually hypo- or isointense to gray matter on T1-weighted sequence and iso- to hyperintense on T2-weighted sequence (Fig. 3.54). Hyperintensity on T2-weighted sequence is attributed to dysplastic or gliotic neural tissue and/or CSF. The mass may appear entirely or partly cystic. Enhancement is unusual but may occur if there is significant vascularity or choroid plexus. Sagittal or coronal MR sequences are important to help distinguish nasal neuroglial heterotopia from a cephalocele by showing a lack of communication between the mass and the brain.127 On CT, the mass is isodense to brain parenchyma or variably hypodense. CT is useful for demonstrating associated bony deformity.
 FIGURE 3.54 Nasal neuroglial heterotopia in a 21-day-old boy who presented with right nasal mass. A: Sagittal postcontrast T1-weighted MR image demonstrates a nonenhancing hypointense mass (arrow) in the nasal cavity. B: The mass (arrow) is predominantly hyperintense on T2-weighted MR image. |
Fissural Cysts
Fissural cysts are cysts that arise along lines of fusion of embryologic facial processes. They include nasolabial cysts, nasopalatine duct cysts, and median palatal cysts.
Nasolabial cyst, also known as nasoalveolar cyst or Klestadt cyst, occurs at the base of the nasal ala and anterior nasal fold and may be bilateral.128 Its density on CT and signal on MR vary depending on the cyst contents,129 which may range from serous to mucous to complex if there is superimposed infection. Enhancement is variable.130,131 When the lesion is large, scalloping of the adjacent bone may be seen (Fig. 3.55).
Nasopalatine duct cysts or median anterior maxillary cysts may be further classified as incisive canal cysts or palatine papilla cysts. The latter are uncommon. The former are thought to arise from nasopalatine duct epithelial remnants.132 Small incisive canal cysts are not uncommon in children. On CT, the cyst appears as a symmetrical pear-shaped, heart-shaped, or rounded lucent area with a sclerotic rim in the midline of the anterior primary palate (Fig. 3.55B). They develop around vital teeth and can cause splaying of the upper central incisor roots.
A median palatal cyst arises at the junction of the palatal processes in the midline. On CT, it appears as a midline lucent lesion in the hard palate.
Clefts
Clefting results from abnormal development or fusion of facial processes.133
Isolated cleft secondary palate: results from insufficiency of the frontonasal and nasomedial processes or lesions
that prevent palatal fusion. Fusion of the secondary palatal shelves is prevented by congenital intraoral masses or severe micrognathia. Underdevelopment of the mandible causes backward and upward displacement of the tongue (glossoptosis). This in turn results in a U-shaped cleft of the secondary palate.

FIGURE 3.55 Fissural cysts. A: Nasolabial cyst. Axial CT image shows a proteinaceous cyst at the base of the left nasal ala. Note flattening and scalloping of the underlying frontal process of the maxilla (arrow). B: Nasopalatine duct cyst. Axial bone window CT image shows a symmetric, expansile, rounded, lucent lesion (arrow) at the distal nasopalatine duct in the midline.
True midline cleft lip and palate: results from failure of fusion of the nasomedial processes. There is hypertelorism when associated with skull base dehiscence and basal cephaloceles. Variable associated hypoplasia or absence of midline structures of the face with hypotelorism should prompt a search for holoprosencephaly.
Common cleft lip and/or cleft palate: these off-midline clefts result from failure of the nasomedial processes to fuse with the maxillary processes. Bilateral cleft lip and palate is associated with a higher incidence of syndromic anomalies than is unilateral cleft palate or cleft lip.
Oblique facial cleft: results from failure of the nasolateral process to fuse with the maxillary process. The cleft extends from the medial canthus of the eye to the nose. This may be seen in association with bilateral common cleft lip and/or palate.

FIGURE 3.56 Micrognathia. A: Treacher Collins syndrome in a 21-month-old boy. 3D CT image shows bilateral symmetric micrognathia and malar flattening. The zygomatic arches are absent. B: Hemifacial microsomia in a 17-month-old girl. 3D CT image shows underdevelopment of the left hemimandible with absence of the ramus and condyle (long arrow). The left zygomatic arch is severely hypoplastic (short arrow). There is complete atresia of the external auditory meatus.
Transverse facial cleft: also termed “wolf mouth” or macrostomia, the transverse facial cleft results from failure of the maxillary and mandibular processes to fuse. This may be unilateral or bilateral. It may be seen in isolation or may be associated with syndromes such as HFM.
Clefts that do not observe lines of embryonic fusion likely result from amniotic band disruption complex.134 Here, rupture of the amnion leads to generation of fibrous bands that can adhere to and tether parts of the developing fetus. Fetal parts may become disrupted, damaged, or amputated by these bands. Craniofacial deformations are seen in approximately one-third of cases of amniotic band sequence.135
Micrognathia
Micrognathia is an abnormally small mandible whereas retrognathia refers to a receding chin, but these two findings often coexist. Micrognathia can be sporadic or inherited, isolated or syndromic. Micrognathia is a feature of trisomies 13, 18, and 9.136 Symmetric micrognathia with hypoplastic and deficient zygomatic arches, malar flattening, and severe anomalies of the external and middle ears is a feature of TCS (Fig. 3.56A) and Nager syndrome. Mildly asymmetric
micrognathia with external, middle, and inner ear anomalies and branchial apparatus anomalies is the hallmark of branchiootorenal (BOR) syndrome. HFM is characterized by unilateral micrognathia, ipsilateral zygomatic arch deficiency, hypoplastic muscles of mastication, and external and middle ear anomalies (Fig. 3.56B).
micrognathia with external, middle, and inner ear anomalies and branchial apparatus anomalies is the hallmark of branchiootorenal (BOR) syndrome. HFM is characterized by unilateral micrognathia, ipsilateral zygomatic arch deficiency, hypoplastic muscles of mastication, and external and middle ear anomalies (Fig. 3.56B).
Moderate to severe micrognathia is associated with airway obstruction. Mandibular hypoplasia tends to displace the tongue posteriorly and superiorly, which, in turn, prevents normal development of the palate resulting in a palatal cleft, as discussed above. The clinical triad of micrognathia, glossoptosis, and feeding difficulty is known as the Robin sequence, which is a feature of various syndromes, for example, velocardiofacial syndrome and Stickler syndrome.
Imaging of micrognathia is required primarily to assist surgical planning prior to mandibular reconstruction. Axial helical CT images using a low-dose algorithm are used to create multiplanar reconstructed images and a 3D model of the mandible and face.
Infectious and Inflammatory Disorders
Bacterial Sinusitis
The two most common predisposing factors for acute bacterial sinusitis are viral upper respiratory tract infection (URI) and allergy.137 If symptoms are persistent (longer than 10 days), severe (high fever and purulent nasal discharge), or worsening after initial improvement, acute bacterial sinusitis is suggested. Symptoms beyond 30 days suggest subacute or chronic sinusitis.137 The most common pathogens are Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis.138,139 Staphylococcus aureus and anaerobic organisms are seen more frequently in children with severe sinus symptoms or with chronic sinusitis (present for over a year).
The ethmoid air cells are separated by thin bony septations; each air cell drains through an independent ostium into the middle meatus.140 These narrow ostia are readily obstructed by inflamed mucosa. When developed, the frontal sinuses are frequently the conduit for spread of infection to the orbit or brain, the latter related to a rich emissary venous plexus between the posterior frontal sinus mucosa and the meninges. Like the frontal sinus, an infected sphenoid sinus is more prone to the development of intracranial complications.
Pediatric sinusitis occasionally arises as a result of odontogenic infection.141 A key CT finding is a periapical lucency due to a periapical abscess or granuloma that is dehiscent into the floor of the maxillary antrum (Fig. 3.22B). Perforation after dental trauma or a dental procedure may lead to a tract that then epithelializes to form an oroantral fistula (Fig. 3.57). Congenital bone defects and dental cysts may also act as conduits for spread of infection to the maxillary sinus.
Sinus inflammation and congestion causes increased secretions and accumulation of submucosal fluid with venous congestion.142 On CT, inflamed sinus mucosa appears as peripheral soft tissue thickening. On MRI, mucosal secretions and submucosal fluid appear hyperintense on T2-weighted sequences and hypointense on T1-weighted sequences. There is a characteristic pattern of a thin rim of enhancement of the inflamed mucosa and nonenhancement of the underlying submucosal fluid (Fig. 3.58). These are findings of sinus inflammation or congestion and not necessarily sinus infection. Sinus CT and MRI often show mucosal thickening in children who do not have signs or symptoms of sinusitis or an URI.143 Therefore, mucosal thickening and edema seen on imaging should not be reported as “sinusitis” without supportive clinical history. Layering fluid in a sinus (air-fluid level) is more suggestive of an acute infectious sinus process (Fig. 3.58).
Related posts:
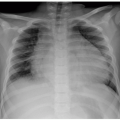
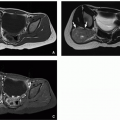
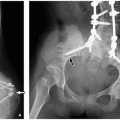
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


