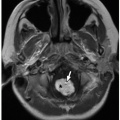
FIGURE 9.1 Standard cardiac magnetic resonance imaging planes. Bright blood magnetic resonance images in (A) two-chamber, (B) four-chamber, and (C) short-axis geometries. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
visible under resting conditions. In general, adenosine is preferred because of its ease of administration, fast action, and short half-life. However, unlike in adults, stress perfusion in pediatrics is usually reserved for those patients clinically felt to be high risk for myocardial ischemia, such as those with coronary artery involvement in Kawasaki disease or in heart transplant patients with suspected coronary artery vasculopathy. Approximately 10 minutes after administration of gadolinium, myocardial viability sequences can be performed to demonstrate retained gadolinium in regions of myocardial fibrosis (Fig. 9.3).
 FIGURE 9.2 Outflow tract cardiac magnetic resonance imaging planes. Bright blood magnetic resonance images in (A) left ventricular outflow tract/three-chamber view (B) right ventricular outflow tract. AA, ascending aorta; LA, left atrium; LV, left ventricle; MPA, main pulmonary artery; RV, right ventricle. |
frequently, general anesthesia or conscious sedation is necessary in the pediatric population.
 FIGURE 9.3 Delayed cardiac wall enhancement. Short-axis plane phase-sensitive inversion recovery magnetic resonance image shows an area of subendocardial enhancement (arrow) in the anterolateral wall of the left ventricle. |
 FIGURE 9.4 Flow quantification using phase-contrast angiography. Phase contrast across the main pulmonary artery (MPA) demonstrates an approximately 40% pulmonary regurgitant fraction (green circle on A and arrow on B). |
on cardiac MRI and is a reliable determinant of ventricular morphology. The tricuspid valve follows the RV. Typically, the tricuspid valve is located very slightly closer to the cardiac apex than the mitral valve. The tricuspid valve has three leaflets, including the septal leaflet (attached to the interventricular septum) and anterior and posterior leaflets. The LV is supplied by the mitral valve and typically has two papillary muscles along its free wall (anterolateral and posteromedial). However, these papillary muscles are more variable in appearance than moderator band in terms of correctly identifying ventricular morphology. If the RV is located anteriorly and rightward relative to the LV, then normal ventricular “D” looping is deemed to have occurred. If the ventricle containing the moderator band is located posteriorly and leftward, then ventricular inversion or “L” looping has occurred. This is discussed in more detail later in the chapter in TGA.
 FIGURE 9.5 Moderator band. Bright blood magnetic resonance image in four-chamber plane shows the moderator band (arrow) in the right ventricle extending from the free wall to the ventricular septum. LV, left ventricle; RV, right ventricle. |
of the AV portion is that it represents one of the boundaries of the triangle of Koch that is the anatomic landmark for the AV node. In addition to the SA node, the AV node forms part of the conduction system of the heart. Along the midportion of the interatrial septum lies a small depression known as the fossa ovalis/foramen ovale that represents the remnant of the ostium secundum and should close shortly after birth.7
 FIGURE 9.6 Normal coronary arteries. A: Axial maximum intensity projection (MIP) CT image demonstrates normal origin of the right coronary artery (RCA) and left coronary artery (LCA) from the right (R) and left (L) sinuses of Valsalva. (N, noncoronary sinus.) B: Three-dimensional volume-rendered CT image shows normal courses of the RCA, LCA, left anterior descending coronary artery (LAD), and left circumflex coronary artery (CIRC). |
inferior aspect of the tricuspid valve annulus. The AV bundle (of HIS) runs through the membranous septum and is the only normal route for connection between the AV node and the ventricular myocardial conduction system.7
 FIGURE 9.7 Diagram of the location of the three types of atrial septal defects. ASD 1: primum atrial septal defects. ASD 2: secondum atrial septal defects. ASD 3: sinus venosus superior/inferior atrial septal defects. |
10% of all ASDs) occur as a result of either malposition of the interatrial septum or malposition of the SVC/IVC resulting in a vena cava that overrides the atrial septum. Therefore, a sinus venous defect is actually a defect above or below the interatrial septum and not truly within the septum like ostium primum and secundum defects. A superior sinus venous defect where the SVC meets the interatrial septum is much more common than the inferior sinus venosus type. The resulting gap between the interatrial septum and the vena cava causes pulmonary venous blood in the left atrium to be drawn into the lower pressure right atrium. In addition, at least 85% patients with superior type sinus venosus defects have concomitant anomalous partial anomalous pulmonary venous drainage from the right upper lobe into the SVC (Fig. 9.9).
 FIGURE 9.8 Ostium secundum atrial septal defect. Bright blood magnetic resonance image in four-chamber plane shows ostium secundum atrial septal defect (arrow). |
 FIGURE 9.9 Sinus venous defect. A: Axial bright blood magnetic resonance (MR) image demonstrates a superior sinus venous defect (asterisk) between the posterior wall of superior vena cava (SVC) and anterior left atrium (LA). B: Three-dimensional volume-rendered MR image shows partial anomalous pulmonary venous return (arrow) from right upper lobe to SVC. |
septal defects (AVSDs) are malformations caused by failure of fusion of the endocardial cushions that result in a deficient AV septum with a common atrioventricular valve (AVV) rather than separate mitral and tricuspid valve openings.14 This common AVV often has five to six leaflets and a complex arrangement of chordal attachments. Normally, the aortic valve is situated between the tricuspid and mitral valves. However, when there is common AVV, the aortic valve becomes displaced superiorly, resulting in an elongated and potentially narrowed left ventricular outflow tract.
 FIGURE 9.10 Balanced atrioventricular septal defect. Frontal chest radiograph shows increased pulmonary vascularity and global cardiomegaly. |
 FIGURE 9.11 Balanced atrioventricular septal defect. Bright blood magnetic resonance image in four-chamber plane demonstrates large atrioventricular septal defect (asterisk). LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
usually close spontaneously, and larger VSDs generally require surgical intervention. Occasionally, the muscular part of the septum can have innumerable VSDs, resulting in the so-called Swiss cheese interventricular septum. Perimembranous VSDs are the most common type to require surgical intervention and are characterized by a defect in the membranous portion of the ventricular septum. The subarterial or malalignment type of VSD appears just below the aortic valve cusps. Perimembranous and subarterial VSDs are often referred to as outlet VSDs and are associated with conotruncal anomalies such as TOF and TGA.17
 FIGURE 9.12 Diagram of the location of the four types of ventricular septal defects (VSDs) including subpulmonary VSD (subarterial), membranous VSD (perimembranous), atrioventricular canal defect (inlet type), and muscular VSD (muscular). |
 FIGURE 9.13 Ventricular septal defect. Frontal chest radiograph shows increased pulmonary vascularity and cardiac enlargement. |
 FIGURE 9.14 Ventricular septal defect. Axial enhanced CT image shows a large muscular VSD (arrow), partially covered by right ventricular muscle bundles. LV, left ventricle; RV, right ventricle. |
 FIGURE 9.15 Perimembranous ventricular septal defect. Black blood magnetic resonance image shows a perimembranous ventricular septal defect (arrow) with muscular extension in a 2-month-old girl with double outlet right ventricle. |
 FIGURE 9.16 Mitral stenosis. Axial bright blood magnetic resonance image shows turbulent jets arising from the tips of stenosed mitral valve leaflets (arrows). LV, left ventricle. |
 FIGURE 9.17 Mitral regurgitation. Two-chamber plane magnitude phase-contrast magnetic resonance image demonstrates mitral valve regurgitation jet (arrow) into an enlarged left atrium (LA). LV, left ventricle. |
 FIGURE 9.18 Mitral valve prolapse. Bright blood magnetic resonance image in left ventricular outflow tract plane shows mitral valve leaflets (asterisks) prolapsing into the left atrium (LA) with a prominent regurgitant jet (arrow). AA, ascending aorta; LV, left ventricle. |
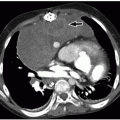
 FIGURE 9.19 Tricuspid stenosis. Axial enhanced CT image shows thickened tricuspid valve with narrowed valve annulus (asterisk) and hypoplastic right ventricle (RV). Also noted is an extracardiac Fontan (F) shunt. |
vascularity1,22 (Fig. 9.23). Echo is used for initial diagnosis. However, MRI is playing an increasing role in the presurgical and postrepair assessment of right ventricular volumes, tricuspid valve morphology/regurgitation, and associated atrial or ventricular shunts.
 FIGURE 9.20 Tricuspid regurgitation. Axial bright blood magnetic resonance image in four-chamber plane shows tricuspid regurgitant jet (arrow) and enlarged right atrium (RA). RV, right ventricle. |
 FIGURE 9.21 Diagram of the anatomy of Ebstein anomaly. The tricuspid valve is downwardly displaced and adherent to the interventricular septum. Part of the right ventricle is “atrialized,” being located superior to the tricuspid valve. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
 FIGURE 9.22 Ebstein anomaly. Bright blood magnetic resonance images in a four-chamber plane (A) and left ventricular outflow tract plane (B) show downward displacement of the attachment of the septal leaflet (straight arrow). The anterior leaflet (curved arrow) is large and “sail-like” resulting in an atrialized portion of the right ventricle (APRV). |
 FIGURE 9.23 Ebstein anomaly. Frontal chest radiograph demonstrates enlarged right cardiac margin. |
aortic valve, small ascending aorta, and tubular hypoplasia of the aortic arch and aortic coarctation (Figs. 9.24 and 9.25).23 If there is severe aortic valve stenosis/atresia, then the ascending aorta and aortic arch may demonstrate retrograde filling from the ductus arteriosus, rather than anterograde flow through the aortic valve. In this situation, the coronary arteries also rely on retrograde flow leading to chronic myocardial hypoperfusion. Most of the oxygenated pulmonary venous blood crosses through an ASD into the pulmonary artery via the right heart and reaches the systemic arterial circulation through the ductus arteriosus.23,24 The descending aorta is usually normal caliber because of the presence of an enlarged ductus arteriosus. Clinical presentation is during the first few weeks of life coinciding with the closure of the ductus arteriosus. Manifestations vary depending on the degree of left-sided obstruction and include failure to thrive, hypoper-fused extremities, cardiac failure, and metabolic acidosis.7,23
 FIGURE 9.24 Diagram of hypoplastic left heart associated with aortic atresia. The left ventricle (LV) is hypoplastic and hypertrophied. The ascending aorta (Ao) is very hypoplastic. LA, left atrium; LPA, left pulmonary artery; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle. |
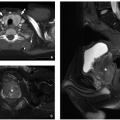
 FIGURE 9.25 Hypoplastic left heart syndrome. A: Axial enhanced CT image shows mitral valve annular stenosis (asterisk) and hypoplastic left ventricle (LV). RA, right atrium; RV, right ventricle. B: Sagittal reformatted enhanced CT image demonstrates tubular hypoplasia of ascending aorta (black arrow) and coarctation (white arrow) at aortic isthmus. |
ventricle palliation is performed,23,24 which results in the RV functioning as the systemic ventricle. This staged complex surgical procedure is performed in three stages, known as the Norwood I, the Glenn shunt (occasionally referred to as superior cavopulmonary anastomosis/Norwood II), and the Fontan shunt (occasionally referred to as Norwood III/total cavopulmonary anastomosis) procedure.23,24
 FIGURE 9.26 Hypoplastic left heart syndrome. Frontal chest radiograph in a 1-day-old girl shows increased pulmonary vascularity and cardiomegaly. Aortic knuckle is not clearly visualized. |
This procedure sacrifices the MPA in order to refashion an ascending aorta capable of sustaining the systemic arterial supply. The MPA is resected and separated from the right and left pulmonary arteries. The MPA is then used to augment the hypoplastic ascending aorta and proximal arch (the Damus-Kaye-Stansel maneuver). As the MPA has been resected, pulmonary arterial supply has to be reestablished. This can be accomplished using either a modified BT shunt from the right subclavian artery to the right pulmonary artery (RPA) (Fig. 9.28) or modified Sano shunt (RV) to confluence of branch pulmonary arteries (Fig. 9.29).23,24
 FIGURE 9.27 Diagram of the Norwood stage 1 operation. The pulmonary artery has been transected at its bifurcation and the distal end oversewn. Pulmonary blood flow is provided by a modified Blalock-Taussig shunt (mBT shunt). The aortic arch is augmented with a patch and connected to the proximal main pulmonary artery. An atrial septectomy is also performed so that pulmonary venous blood can reach the right atrium and then right ventricle without obstruction. LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle. |
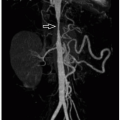
 FIGURE 9.28 Norwood procedure. A: Three-dimensional volume-rendered CT image (A) shows stage 1/modified Blalock-Taussig shunt (asterisk) connecting right subclavian artery (arrow) to right pulmonary artery. B: Coronal reformatted enhanced CT image demonstrates the hypoplastic ascending aorta (arrow), which has been anastomosed to the main pulmonary artery using a Damus Kaye Stansel anastomosis creating the neoaorta (Neo Ao). |
TABLE 9.1 Simplified Description of Selected Named Surgical Procedures for Treatment of Congenital Cardiovascular Anomalies | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
flow returns directly to the pulmonary arteries without going through the heart (total cavopulmonary anastomosis) (Fig. 9.32). There are two types of Fontan shunts currently performed: the lateral tunnel and the extracardiac conduit (Fig. 9.33). In the lateral tunnel Fontan conduit, an intra-atrial baffle is placed in the lateral part of the right atrium connecting the IVC with the undersurface of the RPA. In the extracardiac Fontan conduit, the IVC is transected at the inferior cavoatrial junction. Then, a conduit lying outside the heart is interposed between the IVC and the inferior surface of the RPA.23,24
 FIGURE 9.29 Diagram of the Sano modification for stage 1 palliation of hypoplastic left heart syndrome. A Gore-Tex tube graft connects the right ventricle and pulmonary arteries, provides pulmonary blood flow, and replaces the modified Blalock-Taussig shunt used in the Norwood stage 1 procedure. The Sano modification provides higher systemic diastolic pressures and presumably better coronary perfusion than the Norwood stage 1 procedure. Ao, aorta; LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle. |
 FIGURE 9.30 Diagram of a bidirectional Glenn shunt. The SVC is removed from the heart and connected end to side to the right pulmonary artery (RPA), which is in continuity with the left pulmonary artery (LPA). The main pulmonary artery has been separated from the heart and oversewn. Ao, aorta; Glenn, Glenn shunt; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. |
 FIGURE 9.31 Bidirectional Glenn procedure. Three-dimensional volume-rendered CT image shows stage II/Glenn shunt. Glenn shunt (asterisk) is located between superior vena cava (SVC) and right pulmonary artery (RPA). LPA, left pulmonary artery. |
collateral vessels may also form, which in addition to the venous collaterals, increase preload and risk of dysfunction on the single ventricle. The post-Fontan cardiac MRI assessment involves evaluation of ventricular function as well as systemic veins, pulmonary veins, and pulmonary arteries for evidence of obstruction or thrombosis. In addition, screening for venovenous or aortopulmonary collateral vessels should be performed.28
 FIGURE 9.32 Fontan procedure. Coronal oblique bright blood magnetic resonance image demonstrates Fontan (F) shunt between inferior vena cava (IVC) and right pulmonary artery (RPA) with Glenn shunt (asterisk) as previously placed. |
 FIGURE 9.33 Diagram of an extracardiac Fontan (left) and a lateral tunnel Fontan (right) procedure. The inferior vena cava (IVC) and right pulmonary artery (RPA) are connected by placement of (A) an extracardiac conduit, and (B) a lateral tunnel conduit within the right atrium. In each, a previous bidirectional Glenn shunt (Figure 9.30) has been performed (end-to-side anastomosis of the superior vena vena [SVC] with the RPA). Ao, aorta; LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; RA, right atrium; RV, right ventricle. |
 FIGURE 9.34 Tricuspid atresia. Frontal chest radiograph in cyanotic infant shows mildly decreased pulmonary vascularity, normal heart size, and concavity (arrow) in the region of main pulmonary artery. |
aortic arch (IAA), and aortic coarctation. Pulmonary outflow tract obstruction may also occur with either concordant or discordant ventriculoarterial connections.31 Presentation is usually in the neonatal period with cyanosis and congestive heart failure.
 FIGURE 9.35 Double inlet left ventricle. Axial bright blood magnetic resonance image in four-chamber plane shows both atrioventricular valves (arrows) emptying into the left ventricular chamber (LV) with atretic right ventricle (RV). The right atrium (RA) and left atrium (LA) are also connected with ostium secundum atrial septal defect (*). |
 FIGURE 9.36 Unbalanced atrioventricular canal defect in a 2-month-old girl with heterotaxy. Bright blood magnetic resonance image in four-chamber plane shows large atrioventricular canal defect (AVC) positioned over the right ventricle (RV) (i.e., right-sided dominance) resulting in hypoplastic left ventricle (LV). Also noted is azygos continuation (AZ) of IVC. LA, left atrium; RA, right atrium. |
pulmonary atresia. Absence of the pulmonary valve may also occur in TOF, and this entity is discussed separately later in this chapter. The variable degree of RVOT/pulmonary valve stenosis results in variations in branch pulmonary artery anatomy. In most cases, the pulmonary arteries are normal or mildly diminished in size. However, with pulmonary valve atresia, the branch pulmonary artery anatomy may be very complex. The branch pulmonary arteries may be connected to each other (confluent) and solely supplied either by the ductus arteriosus or by major arteriopulmonary collaterals (MAPCAs).38,39 Nonconfluent branch pulmonary arteries may be solely supplied by multiple MAPCAs. MAPCAs may also supply individual segments of the lung directly without going through the pulmonary arterial branches.
 FIGURE 9.37 Diagram of anatomy of tetralogy of Fallot with right ventricular outflow tract (RVOT) and pulmonary valvar stenosis. The aortic valve (AoV) can be seen through the ventricular septal defect (VSD). There is RVOT stenosis in the subpulmonary valve region, hypoplastic pulmonary valve (PV), hypoplastic pulmonary arteries. A right aortic (Ao) arch is present in ˜25% of patients. LA, left atrium; LV, left ventricle; MPA, main pulmonary artery; RA, right atrium; RV, right ventricle.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|