Hematologic Bone Diseases
Vascular conditions affecting bone may manifest through alterations in blood supply or changes in skeletal architecture caused by abnormal activation of the hematopoietic potential within bone marrow. Unfortunately, the slow metabolic rate of bone significantly delays visualization of quantitative, qualitative, and morphologic changes in the skeleton on low-tech imaging. Advanced imaging techniques such as radionuclide scintigraphy (bone scan) and magnetic resonance imaging (MRI) are capable of demonstrating early changes in bone metabolic rate and microarchitecture.1 The limited number of skeletal responses to hematologic alterations often impairs a physician’s ability to render a specific diagnosis based on plain film alone. In general, vascular conditions of the skeleton have a chronic history of development and nonspecific radiographic findings. Consequently, all radiographic clues must be correlated with historic and laboratory findings. Typically the clinical findings suggest the correct diagnosis long before plain film radiographs are useful.
Avascular Necrosis
Background
Avascular necrosis is the most common hematologic condition affecting the skeleton. Pathologically, this condition is simply bone death resulting from inadequate blood supply. Avascular necrosis is more likely to develop in locations where the blood supply to bone is tenuous or little collateral circulation is present. Many conditions and disorders may be responsible for interrupting blood supply to a segment of bone.11,36
Conditions known to cause avascular necrosis include trauma, alcoholism, infections, medications (including corticosteroids, bisphosphonates, and highly active antiretroviral therapy), dysbaric trauma, marrow infiltration disorders, hypercoagulability states, autoimmune diseases, and idiopathic etiology.4,27,31 These causes may be divided into three categories: (a) conditions resulting in external blood vessel compression near or within the bone, (b) disorders resulting in blood vessel occlusion because of thickening of the vessel wall, and (c) disorders resulting in blood vessel blockage from a thromboembolic process (Box 11-1).12
For conditions resulting in external blood vessel compression, the mechanism is either marrow edema causing compression of the vessel in an enclosed region or excessive packing of the marrow through the deposition of abnormal tissue or material (e.g., fat in steroid administration or hyperlipidemia).16,22,47 Box 11-2 lists, in descending order of frequency, the common causes of ischemic necrosis in children and adults.
Bone ischemia may have several presentations. Infarction affecting a focal segment of the articular surface is termed osteochondritis dissecans in the growing skeleton. This defect may occur in adults and children, and more frequently affects weight-bearing bones at their articular surfaces.19 The lateral aspect of the medial femoral condyle is the most common location of the infarction, followed by the talar dome. Trauma is believed to be the precipitating mechanism, although patients often report no history of trauma. There is evidence to suggest that transient “bone marrow edema syndrome (BMES)” precipitates compression of susceptible vascular structures, occluding the vessels and creating osteonecrosis. 37
The term medullary bone infarction is used to describe ischemic necrosis localized to the medullary portion of a long bone. This type of bony infarction generally is not of primary therapeutic interest because it causes no significant symptoms and is sufficiently distant enough from articular surfaces that it results in no alterations in osseous shape or integrity. Although the finding itself is of no therapeutic importance, medullary bone infarctions are associated with metabolic conditions, such as alcoholism, diabetes, and chronic renal disease, all of which should be ruled out in its presence.
An infarction affecting the entire epiphysis of a skeletally immature long bone is termed epiphyseal ischemic necrosis. The proximal femur is by far the most common location for this event.29 When the proximal femur is affected, the condition is sometimes termed Legg-Calvé-Perthes disease (Box 11-3) in the child or Chandler disease in the adult. Early diagnosis of Legg-Calvé-Perthes disease is important in prevention of postischemic deformity and functional impairment.28 Most cases of Legg-Calvé-Perthes disease have an idiopathic origin, although slipped femoral capital epiphysis, trauma, developmental dysplasia of the hip, and other pathologies also are associated with this condition.
Although ischemia of any segment of a bone is possible, some locations are more likely to be affected because of their vascular supply (Table 11-1). An avascular etiology has been ruled out for some epiphyseal conditions previously attributed to an avascular origin (Table 11-2, Fig. 11-1).
TABLE 11-1
COMMON LOCATIONS FOR ISCHEMIC NECROSIS
| Location | Disorder |
| Metatarsal head | Freiberg disease |
| Tarsal navicular | Köhler bone disease |
| Talus | Diaz disease |
| Patella (secondary | Sinding-Larsen-Johansson ossification center) disease |
| Medial femoral condyle | Spontaneous osteonecrosis of the knee (SONK) |
| Femoral head (child) | Legg-Calvé-Perthes disease |
| Phalanges of hand | Thiemann disease |
| Metacarpal head | Mauclaire disease |
| Carpal lunate | Kienböck disease |
| Carpal scaphoid | Preiser disease |
| Capitellum of the humerus | Panner disease |
| Humeral head | Hass disease |
| Vertebral body | Kümmel disease |
TABLE 11-2
EPIPHYSEAL CONDITIONS UNRELATED TO AVASCULAR NECROSIS
| Condition | Etiology | Description |
| Blount disease (tibia vara) | Trauma | Local growth alteration of the medial portion of the proximal tibial epiphysis; infantile and adolescent presentation; depressed medial metaphysis of the tibia with an osseous overgrowth noted; shortening of involved leg; usually a tibia vara deformity |
| Osgood-Schlatter disease | Trauma | Altered appearance of the tibial tuberosity occurring mostly between the ages of 11 and 15 years; more common among boys and girls who participate in sports such as soccer and weight lifting; fragmentation and soft-tissue swelling of the tibial tuberosity evident on radiographs; pain and tenderness over the region; tibial tuberosity possibly fragmented as a normal variant and distinguishable from Osgood-Schlatter disease by lack of pain and soft-tissue swelling |
| Scheuermann disease* | Trauma | Posttraumatic defect of vertebral endplate maturation first seen during adolescence with three or more levels of wedged vertebrae, narrowed anterior disc space, multiple Schmorl nodes, and vertebral endplate irregularity; middle and lower thoracic spine are the usual locations; back pain is common; more severe kyphosis possibly necessitates bracing or, in a few cases, surgery to arrest or partially correct the resulting deformity |
| Sever phenomenon | Variation in ossification | Irregularity and fragmentation of the secondary ossification center of the calcaneus; generally considered a normal variant and unrelated to any heel pain in the adolescent |
| Sinding-Larsen-Johansson disease | Trauma | Fragmented appearance of the lower pole of the patella, most commonly occurring between 10 and 14 years of age; soft-tissue swelling and tenderness common and exacerbated by activity |
The histologic changes in bone necrosis are essentially the same, regardless of location. Pathologically, absence of a blood supply results in bone death within 48 hours; a predictable sequence of events then ensues: inflammatory reaction to the dead bone, neovascular infiltration into the dead segment of bone, resorption of dead bone and deposition of new bone, and remodeling of the resultant bone.30 This process may take 1 to 8 years to complete.
Imaging Findings
Osteochondritis Dissecans.
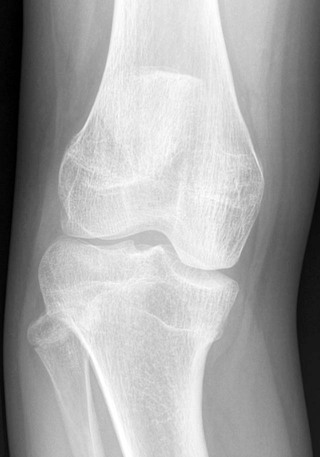
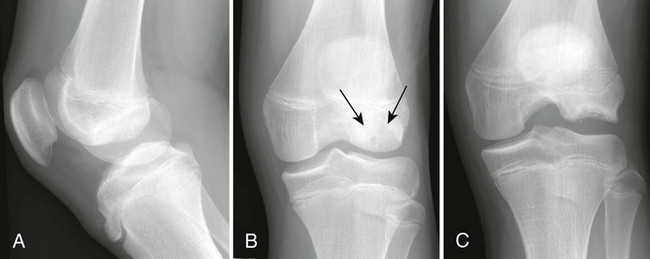
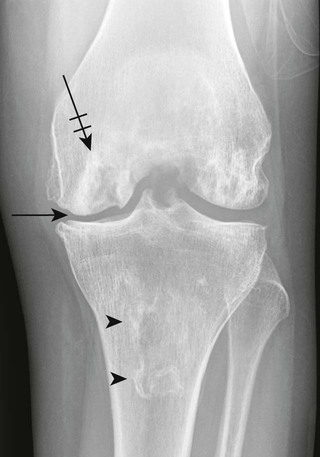
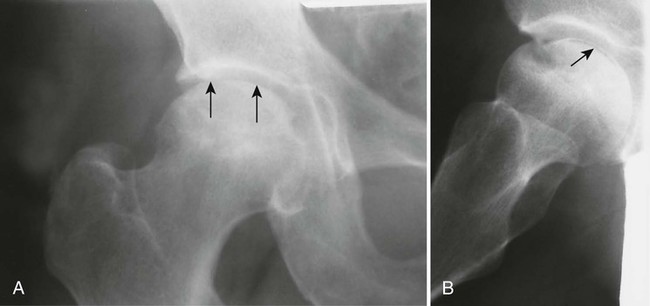
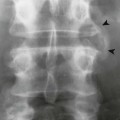
Osteochondritis dissecans (focal subarticular infarction) generally first manifests radiographically, the plain film demonstrating a subarticular lucent defect accompanied by a free-floating fragment of bone that formerly filled the defect. The defect is characteristically smooth and regular with varying degrees of marginal sclerosis corresponding roughly to the shape of the fragment (Fig. 11-2). The fragment may be displaced from the parent bone defect, or remain in in situ. The marginal sclerosis around the defect is most pronounced at the margin of the defect, fading gradually into the subchondral bone peripherally (Fig. 11-3). These findings assist in differentiation of focal subchondral infarction from an acute intraarticular fracture. Intraarticular swelling is often noted, but its severity varies considerably (Figs. 11-4 and 11-5).
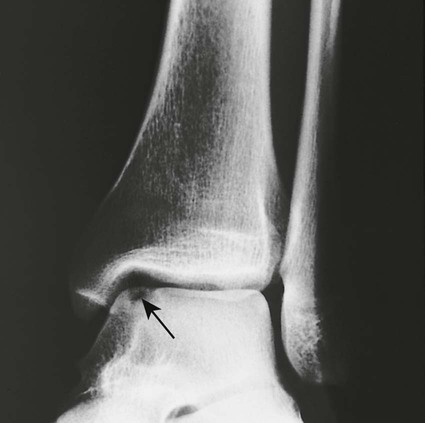
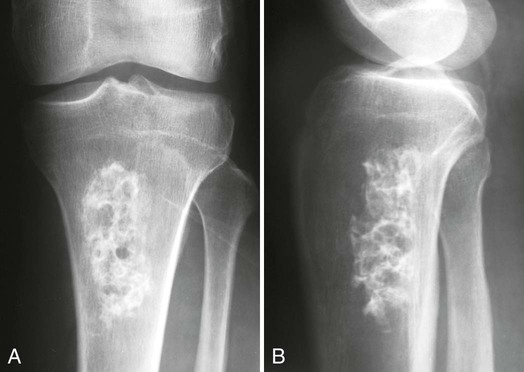
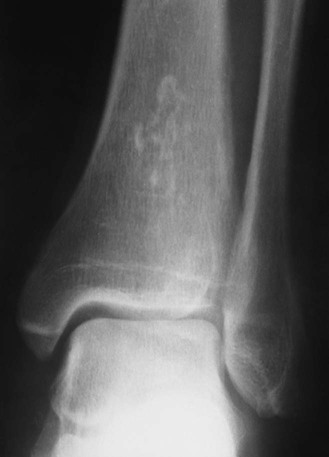
Medullary Bone Infarction.
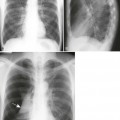
Plain film radiographs are not capable of demonstrating the acute manifestations of a medullary bone infarction. MRI and scintigraphy are demonstrative of the pathology within days of its occurrence. Weeks to months later, after resolution and revascularization, the infarction is usually visible radiographically as discontinuous, longitudinally oriented, and wavy or serpiginous calcific opacities lying centrally in the medullary cavity (Figs. 11-6 to 11-8). The infarction does not result in periosteal reaction, alteration in the cortical thickness, or expansion of the external architecture of the affected bone. Differentiating a medullary bone infarction from a benign enchondroma of bone can be impossible on plain film images. However, definitive differentiation generally is unnecessary because both conditions are benign.
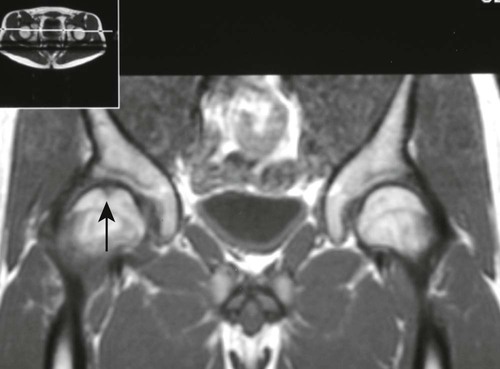
Epiphyseal Necrosis.
Avascular Phase.
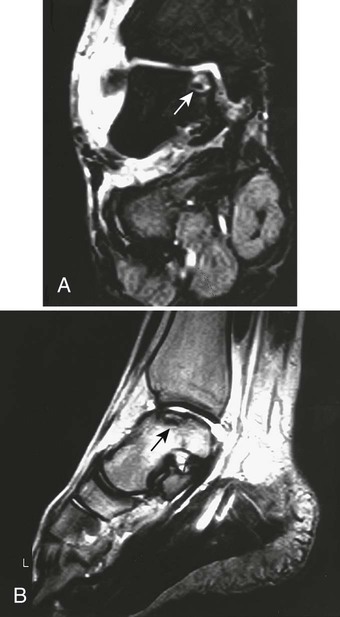
From both therapeutic and prognostic standpoints, epiphyseal necrosis is a significantly more important condition than medullary bone infarction. During the avascular phase of epiphyseal necrosis, plain film radiographic skeletal changes are absent (Fig. 11-9).24 Intraarticular effusion may be visible, but the degree of effusion varies, making it an unreliable indicator of the condition.
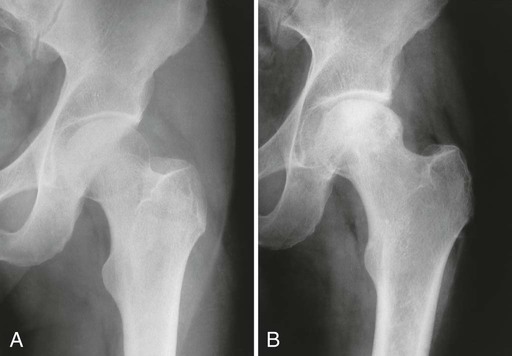
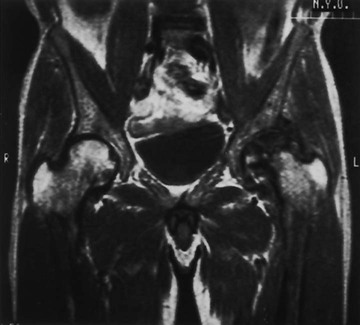
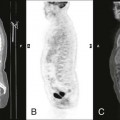
Changes in marrow signal can be identified with MRI, which is the most sensitive imaging modality for the detection of early bone infarction.5 MRI demonstrates loss in the epiphyseal marrow signal, particularly on T1-weighted images, even in the earliest phases of the disease (Fig. 11-10).10,18,46
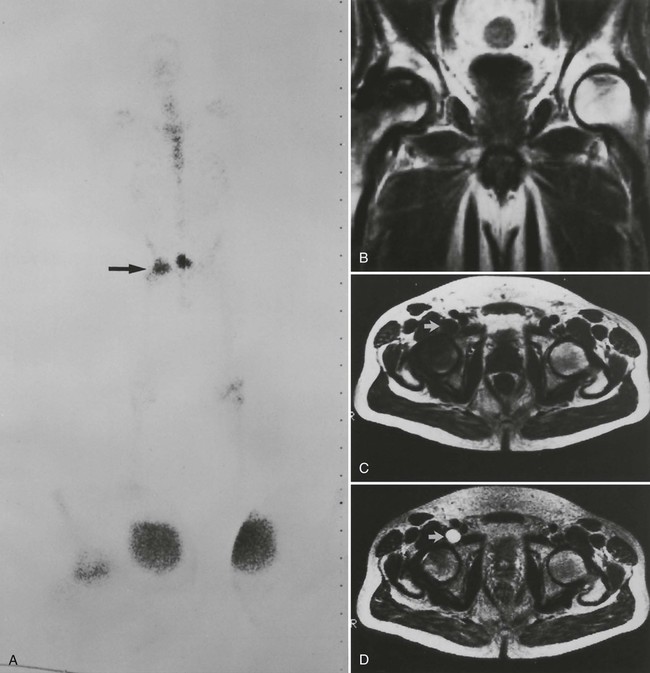
Early epiphyseal infarction also can be detected by radionuclide scintigraphy, which demonstrates focal photopenia affecting the avascular segment. An ill-defined zone of increased radionuclide uptake is present in the region surrounding the avascular segment, presumably resulting from reactive synovitis and hyperemia (Fig. 11-11).9
Inflammatory Phase.
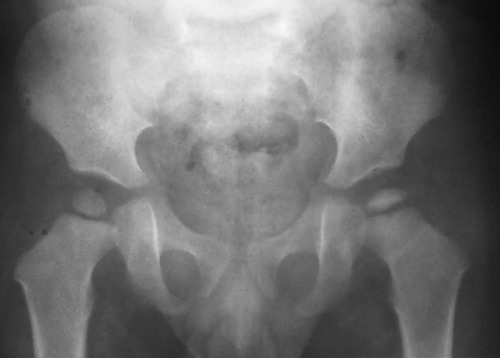
As the avascular phase gives way to the inflammatory response, plain films may demonstrate periarticular osteopenia affecting all but the involved segment of bone. This may create the visual illusion of sclerosis of the avascular segment. This subtle finding, like so many other early plain film signs is not an entirely reliable indicator of necrosis. (Fig. 11-12). The inflammatory phase may persist for weeks.
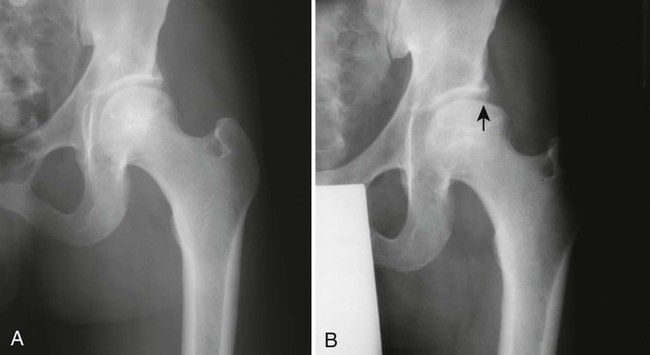
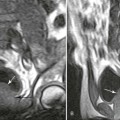
During this time, gross architectural changes to the dead bone begin to develop. The stricken bone softens and may collapse under the stresses of normal use. With this collapse, the involved articular surface becomes deformed (Figs. 11-13 and 11-14). Subchondral fractures that interrupt the cartilaginous cap of the bone allow joint fluid to intrude into the subchondral ischemic bone. This fluid intrusion may produce a subchondral lucency defect in the subchondral bone paralleling the subchondral cortical surface. This arciform lucency is known as the “crescent sign” on plain films (Fig. 11-15). The crescent sign is considered the most reliable early plain film sign of epiphyseal infarction.24 Larger regions of bony collapse may develop, appearing as semilunar radiolucent defects extending from the articular cortex. The defects have been likened to configuration of a bite mark, known as the “bite sign.” The Gage sign is a variation of the bite sign and is visualized as a radiolucent defect of bone (often shaped as a V) appearing at the lateral margin of the involved epiphysis. As this series of gross architectural changes occurs, the healing response begins. Articular collapse is common (see Fig. 11-20).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


