- •
Identify the location of the abdominal organs (stomach and liver) and determine situs.
- •
Identify the position of the heart in the chest (levocardia or dextrocardia).
- •
Identify the systemic venous connections (is there a right superior vena cava? Is there a left superior vena cava? Does the inferior vena cava insert into the heart?).
- •
Look for the presence of a dilated azygos vein at the level of the diaphragm, which suggests interruption of the inferior vena cava and azygos continuation of inferior systemic venous return.
- •
Identify the nature of the hepatic venous return and its connections to the atria.
- •
Identify the presence or absence of the atrial septum (is there a common atrium?).
- •
Identify the presence or absence of two relatively equal-sized ventricles.
- •
Look for ventricular septal defects, size, and location.
- •
Identify the atrioventricular valve connections in relation to the ventricles.
- •
Identify the presence and degree of atrioventricular valve insufficiency.
- •
Identify the position and origin of the great vessels.
- •
Determine the nature of any outflow tract obstruction to the pulmonary artery or aorta.
- •
Assess the relative size of the pulmonary artery to the aorta.
- •
Assess aortic arch anatomy and size.
- •
Assess main and branch pulmonary artery anatomy and size.
- •
Identify the direction of flow across the ductus arteriosus, as either antegrade from pulmonary artery–to–aorta or retrograde from aorta–to–pulmonary artery.
- •
Determine the pulmonary venous connection and drainage to the heart.
- •
Search for any venous structure in the mediastinum or liver in which there is Doppler flow suggesting blood flow away from the heart, because this is highly suggestive of the presence of anomalous pulmonary venous drainage.
- •
Look for rhythm irregularity that may suggest the presence of heart block.
Anatomy and Anatomical Associations
Heterotaxy is of Greek origin, with heteros meaning “other” and taxis meaning “arrangement.” Heterotaxy syndrome is a disorder of abnormal lateralization of the abdominal viscera, thoracic organs, and cardiac atria. Visceral location falls in a spectrum between situs solitus (normal) and situs inversus (inverted), and cardiac position may vary from levocardia (heart positioned on the left side) to dextrocardia (heart positioned on the right side). In addition to these findings, organs that usually manifest characteristic right-left asymmetry such as the lungs and atrial appendages develop in a mirror-image way. Heterotaxy syndrome can generally be categorized into two subtypes, polysplenia (also known as left atrial isomerism ) and asplenia (also known as right atrial isomerism ), although some patients exhibit features of both. Complex congenital heart disease is a hallmark of both subtypes; in addition, many patients have functional asplenia and malrotation of the intestines. Heterotaxy syndrome results in high morbidity and mortality among children with congenital heart disease despite surgical advances in the current era. The diagnosis is readily made in fetal life when particular cardiac defects are seen in combination or when pathognomonic features of the disease such as organ displacement and organ symmetry are observed.
Heterotaxy syndrome can have a spectrum of congenital heart defects, the hallmarks being systemic and pulmonary venous anomalies, inlet abnormalities, and conotruncal defects.
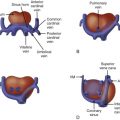
In asplenia , there is bilateral right-sidedness. The atrial appendages both typically have the morphological features of a right atrial appendage (broad-based, triangular shape). There is often absence or unroofing of the coronary sinus and dual sinus nodes. Specific characteristics define the right and left lungs. Normally, the right lung has three lobes, and the left two. In the right lung, the bronchi are considered “eparterial,” which means the airway sits on top of, or above, the arteries as they run in parallel into the parenchyma, and in the left lung, the bronchi are “hyparterial,” which means the airways sit beneath the arteries. In asplenia, regarding the lung anatomy, there are usually two trilobed (right) lungs with bilateral eparterial bronchi. The systemic and pulmonary venous systems are often abnormal with bilateral superior venae cavae and total anomalous pulmonary venous connection (TAPVC, Figure 28-1 ), frequently to an extracardiac site. The inferior vena cava is often juxtaposed to the descending aorta adjacent to the spine; and hepatic venous drainage may be contralateral to the inferior vena cava because it can enter the atria separately. Complex congenital intracardiac disease is a common feature and typically includes common atrioventricular canal (AVC) in association with a conotruncal abnormality such as transposition of the great arteries or double-outlet right ventricle (DORV). Pulmonary outflow obstruction including pulmonary atresia is usual, whereas systemic outflow obstruction is rare.

In polysplenia, bilateral left-sidedness is seen. Both atrial appendages tend to have left appendage anatomy (a long, tubular appearance with a narrow neck). The sinus node may be absent or hypoplastic (because it is typically a right atrial structure). The lungs both have left lung features with two lobes and hyparterial bronchi. The systemic and pulmonary venous connections are also frequently abnormal. Interruption of the hepatic portion of the inferior vena cava with azygos continuation to the superior vena cava is a pathognomonic finding of polysplenia and occurs in approximately 60% to 90% of cases. More than 50% of patients with polysplenia also have bilateral superior venae cavae. With regard to the pulmonary veins, TAPVC to an extracardiac site has been reported in only one case. However, ipsilateral pulmonary veins, in which the right pulmonary veins enter the right atrium and the left pulmonary veins enter the left atrium, is quite common. This occurs in up to 60% of cases, typically in association with a large common atrium with no identifiable atrial septum. TAPVC directly to the right atrium can also occur particularly when there is predominantly situs inversus. Intracardiac abnormalities can be less severe than those seen in asplenia. Approximately 15% of those with polysplenia may have normal intracardiac anatomy with interruption of the inferior vena cava as the only feature. When structural heart disease is present, typical cardiac abnormalities include common AVC, common atrium, single ventricle, and pulmonary outflow obstruction. In some cases, left-sided obstructive lesions and hypoplastic left heart syndrome can be seen.
In both asplenia and polysplenia, dextrocardia and right aortic arch with mirror image branching occur approximately one third of the time. Up to 20% of both subtypes have absence of the right superior vena cava with isolated left superior vena cava.
Splenic abnormalities including complete absence of the spleen (which can occur in asplenic and polysplenic subtypes) or small, multiple spleens are common in heterotaxy syndrome. In association with the anatomic abnormalities, splenic function may be normal, marginal (polysplenia), or nonexistent (asplenia). Those with splenic dysfunction are vulnerable to particular bacterial infections. The liver is often midline rather than right or left sided. Intestinal malrotation, another common association, affects up to 30% to 40% of children with heterotaxy syndrome and is not readily identified on fetal examination. Biliary atresia has been reported in 3% to 10% of heterotaxy patients and is more common in those with polysplenia. Midline defects such as cleft palate and neurological, musculoskeletal, and genitourinary abnormalities have also been reported. Complete heart block is also unique to patients with polysplenia and carries a very poor prognosis.
Frequency, Genetics, and Development
It is difficult to determine the frequency of heterotaxy syndrome because many different cardiac lesions are seen in association with the syndrome and it is frequently underdiagnosed. It is considered a relatively rare abnormality but accounts for significant morbidity and mortality in most large pediatric cardiac care centers.
Several genes including ZIC3 and CRYPTIC have been associated with heterotaxy syndrome; these genes are likely involved in the regulation of signaling of early embryological left-right patterning. Several mice models including the inversum viscerum have abnormalities of left-right asymmetry with mutation of the Lrd gene. Many of these mice models also have abnormalities of nodal cilia suggesting features similar to Kartagener’s syndrome.
Asymmetrical expression of signaling molecules, including Sonic hedgehog, cNR1, Lefty-2, and Pitx2 are involved in the development of heterotaxy syndrome. Many of these have had direct mechanistic implications for the development of asymmetrical structures. Nodal cilia in most vertebrates rotate rapidly to produce flow from right to left, a mechanism now identified as the initial event in breaking left-right symmetry. In heterotaxy syndrome, this mechanism appears to be affected very early in development, resulting in either bilateral right- or bilateral left-sidedness.
Prenatal Physiology
The physiology of heterotaxy syndrome as it presents in utero is dependent on the cardiac anatomy. A systematic assessment of the fetal heart and vasculature is necessary in order to diagnose heterotaxy syndrome. This requires knowledge of the segmental approach to anatomical diagnosis of congenital heart disease, as established by Anderson and coworkers and Van Praagh and colleagues. In an orderly, sequential manner, this approach addresses cardiac position, systemic and pulmonary venous anatomy, atrioventricular (AV) connections, ventriculoarterial connections, relationship of the great arteries to each other, and aortic arch anatomy. All of these aspects of cardiac anatomy can be abnormal in heterotaxy syndrome.
The diagnosis of heterotaxy syndrome is suggested on fetal echocardiogram when many typical features are seen in combination. Determination of fetal position is essential and should be performed early in the scan so that cardiac position and location of abdominal organs such as the liver and stomach may be correctly identified. Discrepancy between cardiac position and location of visceral structures (i.e., levocardia with stomach on the right or dextrocardia with stomach on the left) should alert the practitioner to the possibility of heterotaxy syndrome. Approximately 20% to 40% of those with heterotaxy syndrome have been reported to have dextrocardia with slightly higher prevalence in those with asplenia.
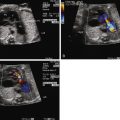
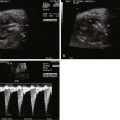
Once fetal position and lie are established, the coronal view of the abdomen can identify whether there is interruption of the inferior vena cava (as is seen in polysplenia). If interruption is present, the coronal view will identify two circles side-by-side in the retroperitoneal region just anterior to the spine, one being the aorta and the other the enlarged azygos vein. In the longitudinal view, two vessels can be seen in parallel with flow in opposite directions by color Doppler. The azygos will have a venous flow pattern and be directed toward the heart, and the aorta will be pulsatile and directed away from the heart. No intrahepatic portion of the inferior vena cava can be identified when there is interruption of the inferior vena cava. In asplenia, the inferior vena cava is usually connected to the right atrium (or the left atrium) and can be demonstrated in the longitudinal view, but it is often juxtaposed to the aorta. Other components of the systemic venous anatomy can be identified including presence or absence of the right superior vena cava, assessment for bilateral superior venae cavae, and the connection of the left superior vena cava to the heart (i.e., directly into the roof of the left atrium or to the coronary sinus).
The pulmonary venous anatomy must be determined ( Figure 28-2 ). Functional single ventricle in association with extracardiac TAPVC carries a very poor prognosis. Therefore, identification of TAPVC is important for family counseling and for postnatal management. In contrast to other congenital heart defects, obstructed TAPVC is a surgical emergency with maintenance of ductal patency providing little to no assistance in stabilizing the postnatal physiology. TAPVC occurs in 60% of patients with asplenia syndrome and should always be highly suspected in this population. Although TAPVC cannot always be identified accurately, the practitioner should look for abnormal venous structures posterior to the heart that may be carrying the pulmonary venous blood flow to an extracardiac site such as the innominate vein, superior vena cava, left superior vena cava, or below the diaphragm into the liver.

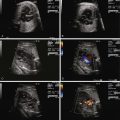
In heterotaxy syndrome, particularly in those with asplenia, the AV connection is typically an AVC defect (see Chapter 9 ). The unbalanced form of AVC occurs frequently in heterotaxy, with one viable ventricle present. In the short-axis view, the common AV valve can often be seen en face. Because the common AV valve can be structurally abnormal, in utero regurgitation may occur. Color flow mapping readily identifies fetal AV valve regurgitation and its severity. If the AV valve regurgitation is severe, volume overload develops. Persistence of severe regurgitation often results in fetal compromise with heart failure and the development of hydrops fetalis.
The ventriculoarterial connections can also be abnormal in heterotaxy syndrome. Conotruncal defects typically seen include transposition of the great arteries, DORV, and right ventricle–to–aorta with pulmonary atresia. The prevalence of these lesions ranges from 50% to 96%, with a higher frequency in asplenia than in polysplenia. These conotruncal anomalies are usually seen in association with common AVC defect so that when both are seen together, heterotaxy syndrome should be highly suspected. On fetal echocardiography, both great vessels should be identified (if present) with determination of their connections to the ventricles. In transposition of the great arteries, the great vessels will be in parallel rather than cross each other. In DORV, blood from the left ventricle (if present) must pass through a ventricular septal defect (VSD) to get to the great vessels. In some cases, one great vessel overrides the VSD. In right ventricle–to–aorta, the aorta arises directly from the right ventricle and the pulmonary valve cannot be identified because there is atresia. Typically, the pulmonary arteries receive blood flow retrograde from the ductus arteriosus, which has a tortuous appearance.
Pulmonary outflow obstruction, either pulmonary stenosis or the more severe pulmonary atresia, is also observed in the majority of patients with asplenia and approximately half the patients with polysplenia. Retrograde flow in the ductus arteriosus heralds significant right ventricular outflow tract obstruction. Systemic outflow obstruction is more common in polysplenia, with hypoplastic left heart syndrome seen on occasion.
Complete heart block can be seen in association with the polysplenic subtype of heterotaxy syndrome. This finding alerts the obstetrician to refer for cardiac evaluation because the ventricular rate may be significantly less than normal, often in the range of 50 to 70 bpm. When the ventricular rate is very low, hydrops fetalis can occur.
Prenatal Management
Because most patients with heterotaxy syndrome have complex congenital heart disease, close follow-up is required during the course of gestation. Risk factors for poor outcome include a diagnosis of functional single ventricle in association with TAPVC, significant AV valve regurgitation, or complex heart disease with complete heart block. Fetal demise may occur, particularly in those with complete heart block or hydrops fetalis due to valve regurgitation. Families may consider termination of the pregnancy based on severity of the cardiac disease. If the pregnancy is continued, monthly follow-up echocardiographic studies are suggested to follow progression of disease. In those with pulmonary stenosis, serial echocardiography may identify progression to pulmonary atresia. In cases with minimal or no AV valve regurgitation and no complete heart block, the disease may remain quiescent during gestation with no significant physiological impact on the fetus or the mother.
Direction and restriction of flow at the atrial septum (if there is functional single ventricle such as hypoplastic left heart syndrome or tricuspid atresia) and the ductus arteriosus should be performed at each monthly assessment to help to establish the severity of disease. Serial studies also allow for continued consultation with the family and discussion about any changes that may have taken place.
One of the most important aspects of determining prognosis for the fetus with heterotaxy syndrome is knowing the anatomy and drainage of the pulmonary veins. At initial evaluation, for example, at 18 to 22 weeks, visualization of the pulmonary veins can be extremely challenging, because there is normally very little flow in the pulmonary circulation at this gestational age. Compounding the difficulty may be the presence of pulmonary stenosis further limiting pulmonary blood flow or pulmonary venous obstruction due to anomalous connection. Serial evaluation over time, in particular as the fetus approaches later in the third trimester, at a time when pulmonary blood flow is relatively increased in comparison to the second trimester, may help identify pulmonary venous connection not previously seen.
For those fetuses with complete heart block in association with heterotaxy syndrome, the prognosis is quite poor. In a series of 11 such patients who received aggressive treatment after birth including early pacemaker implantation, there were no survivors. Many of these fetuses are noted to have abnormal-appearing myocardium that may affect outcome. Families carrying fetuses with this combination of defects benefit from being counseled accordingly.
From a cardiac standpoint, cesarean section is not necessary unless there is (1) concern about heart block or (2) concern about obstructed TAPVC, both of which may be a surgical emergency at birth. In those cases, urgent delivery with rapid transition to a cardiology and cardiac surgery team may result in the best outcome. If risk factors are not present, normal labor and delivery is optimal for the transition. As with any significant cardiac defect, premature delivery increases the risk of poor outcome because of the additional potential comorbidities such as lung disease, retinopathy, neurological injury, and necrotizing enterocolitis.
Postnatal Physiology
The clinical presentation of newborns with heterotaxy syndrome varies depending on the underlying cardiac anatomy. With prenatal diagnosis, practitioners are prepared for potential hemodynamic instability after birth. Long-term outcome may improve with prenatal diagnosis of many congenital heart lesions ; however, in the case of heterotaxy syndrome, the outcome is often dictated by the severity of the disease. Prenatal diagnosis allows for determination of whether the infant will be ductal dependent (defined as a clinical situation in which the ductus arteriosus must remain open to provide adequate pulmonary or systemic blood flow). In general, accuracy in the prediction of ductal dependency is quite high for heterotaxy syndrome.
Infants with asplenia who have single ventricle with pulmonary stenosis or atresia will be cyanotic at birth. The cyanosis will be severe if obstructed TAPVC is also present because blood will not be able to exit the lungs. For those with normal pulmonary venous connections, initiation of prostaglandin E 1 will provide hemodynamic stability and the mild cyanosis exhibited will be well tolerated. If obstructed TAPVC is present, prostaglandin E 1 may not improve oxygenation because pulmonary venous blood flow is hindered on its return from the heart. Postnatal echocardiography will help in this determination. In this clinical situation, if the partial pressure of oxygen (PO 2 ) remains less than 30 torr despite aggressive medical management, urgent surgical intervention may be required.
In polysplenia, a broader spectrum of congenital heart disease may be seen. Patients with severe right or left ventricular outflow tract obstruction will require prostaglandin E 1 after birth until surgery is performed. In those with less severe forms of heart disease, surgery may not be required in the neonatal period and there may be no symptoms in early postnatal life. In patients who have a large VSD with no significant pulmonary stenosis, congestive heart failure will develop as the pulmonary vascular resistance drops (generally at 6-8 wk of life).
Those patients with complex congenital heart disease and complete heart block may exhibit signs of low cardiac output, particularly if the ventricular rate is less than 55 bpm. In some cases, inotropic medications may be required to augment perfusion. If these measures fail, pacing is required.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


