Nicola Sverzellati, Zelena A. Aziz, David M. Hansell
High-Resolution Computed Tomography of Interstitial and Occupational Lung Disease
The pulmonary interstitium is the network of connective tissue fibres that supports the lung. It includes the alveolar walls, interlobular septa and the peribronchovascular interstitium. The term interstitial lung disease (ILD) is used to refer to a group of disorders that mainly affects these supporting structures. Although the majority of these disorders also involve the air spaces, the predominant abnormality is usually thickening of the interstitium which may be due to the accumulation of fluid, cells, or fibrous tissue.
The chest radiograph remains part of the initial assessment of ILD, but the radiographic pattern is often non-specific, observer variation is considerable and it is relatively insensitive to early ILD.1,2 High-resolution computed tomography (HRCT) has revolutionised the imaging of ILD as it enables early detection of disease, allows a histospecific diagnosis to be made in certain cases, and provides insights into disease reversibility and prognosis.
High-Resolution Computed Tomography Patterns of Diffuse Lung Disease
Before considering the individual HRCT patterns related to each ILD, an understanding of normal lung anatomy is needed. In addition, radiologists should refer to a common terminology outlined in the Fleischner Glossary to describe HRCT abnormalities.3 Diffuse abnormalities of the lung on HRCT may be broadly classified into one of the following four patterns: (A) reticular or linear; (B) nodular; (C) ground-glass opacity through to consolidation; and (D) areas of decreased lung attenuation.
Reticular Pattern
A reticular pattern on CT almost always represents significant ILD. Morphologically, a reticular pattern may be caused by thickened interlobular or intralobular septa or honeycomb (fibrotic) destruction. Numerous thickened interlobular septa indicate an extensive interstitial abnormality and causes include infiltration by fibrosis (interstitial fibrosis), abnormal cells (lymphangitis carcinomatosa), or fluid (pulmonary oedema). Although thickened interlobular septa can be a consequence of infiltration by fibrosis, this feature is not a frequent finding in idiopathic pulmonary fibrosis (IPF). Interlobular septal thickening is usually described as smooth (seen in pulmonary oedema and alveolar proteinosis) or irregular (e.g. lymphangitic spread of tumour), but the distinction is not always easily made. Sarcoidosis causes nodular septal thickening, although this pattern is not usually the dominant feature of parenchymal involvement.4
In some diseases, a perilobular distribution may give the spurious impression of thickening of the interlobular septa. However, such a pattern reflects a pathologic process that is ‘smeared’ around the internal lobular surface and is most frequently associated with organising pneumonia.3,5
Intralobular septal thickening manifests as a fine reticular pattern on HRCT and is seen in all ILDs but most commonly in IPF. Often, the intralobular septal thickening may be so fine that HRCT does not demonstrate discrete intralobular opacities but a generalised increase in lung density (ground-glass opacification). Severe pulmonary fibrosis usually results in a coarse reticular pattern made up of interlacing irregular linear opacities. The reticular pattern of end-stage fibrotic (honeycomb) lung is characterised by cystic air spaces surrounded by irregular walls. The certain identification of honeycombing, as opposed to other forms of reticulation, is not always straightforward but is of particular relevance as it may directly impact patient care.3 The distortion of normal lung morphology by extensive fibrosis results in irregular dilatation of segmental and subsegmental airways (traction bronchiectasis/bronchiolectasis); in the periphery of the lung, it can be difficult to distinguish dilated airways from true honeycomb destruction.
Nodular Pattern
A nodular pattern is a feature of both interstitial and airspace diseases. The distribution and density of nodules may help narrow what can be a lengthy differential diagnosis.6 Nodules within the lung interstitium, especially those related to the lymphatic vessels, are seen in the interlobular septa, subpleural and peribronchovascular regions; this distribution is seen most frequently in sarcoidosis but also in lymphangitis carcinomatosa. Centrilobular nodules are seen in several conditions (Table 16-1). Distinguishing between subacute hypersensitivity pneumonitis and respiratory bronchiolitis–interstitial lung disease can be difficult, because both cause relatively low-density, poorly defined centrilobular nodules which may look identical on HRCT. A random distribution of very small well-defined nodules is seen in patients with haematogenous spread of tuberculosis, pulmonary metastases, pneumoconiosis and rarely sarcoidosis.

Ground-Glass Pattern
A ground-glass pattern on HRCT is defined as a generalised increase in opacity that does not obscure pulmonary vessels.3 At a microscopic level, the changes responsible for ground-glass opacity are complex and include partial filling of the air spaces, considerable thickening of the interstitium, or a combination of the two. Ultimately though, the pattern of ground-glass opacity on HRCT results from displacement of air from the lungs. Indeed, ground-glass opacity may be found in many conditions, either as the predominant or as an ancillary pattern. A predominant ground-glass pattern may be seen in subacute hypersensitivity pneumonitis, acute respiratory distress syndrome (ARDS), acute interstitial pneumonia (AIP), non-specific interstitial pneumonia (NSIP) and some infections, notably viral pneumonias, and Pneumocystis jiroveci.
Mosaic Attenuation Pattern
The term ‘mosaic attenuation pattern’, or more simply mosaic pattern, refers to regional attenuation differences demonstrated on HRCT. The attenuation of a given area of lung depends on the amount of blood, parenchymal tissue and air in that area, and thus the sign of a mosaic attenuation pattern is non-specific. It is the dominant abnormality in three completely different types of diffuse pulmonary disease: small airways disease, chronic occlusive vascular disease and infiltrative lung disease. In the first two processes, the decreased attenuation (‘black’) lung is abnormal; in infiltrative lung disease it is the ‘grey’ lung that is abnormal. In a study of 70 patients in whom a mosaic attenuation pattern was the dominant abnormality, Worthy et al. showed that small airways disease and infiltrative lung disease were correctly identified but the mosaic attenuation pattern caused by occlusive vascular disease was frequently misinterpreted.7 Bronchial abnormalities and, to a lesser extent, the presence of air trapping on expiratory CT are the most useful discriminatory features in identifying small airways disease as the cause of mosaic attenuation. However, the phenomenon of hypoxic bronchodilatation in chronic occlusive vascular disease, and the fact that air trapping may be seen on expiratory CT in chronic thromboembolic disease, complicates interpretation.8 Nevertheless, the differentiation between the three basic causes of a mosaic attenuation pattern is usually easily made when clinical and physiological information is taken into account.
Idiopathic Interstitial Pneumonias
The term idiopathic interstitial pneumonia (IIP) is applied to a group of disorders with no known cause, and with more or less distinct histological and radiological appearances.9 In the updated American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus classification of the IIPs, the overall architecture of the classification is preserved (Table 16-2), but the clinical entity, rather than the histopathological label, is given pre-eminence. In addition, an entity termed idiopathic pleuroparenchymal fibroelastosis (IPPFE) is included.10,11 The authors of the updated classification again recommend a multidisciplinary and dynamic approach to the diagnosis of the IIPs which encourages interaction between clinicians, radiologists and pathologists.
Usual Interstitial Pneumonia/Idiopathic Pulmonary Fibrosis
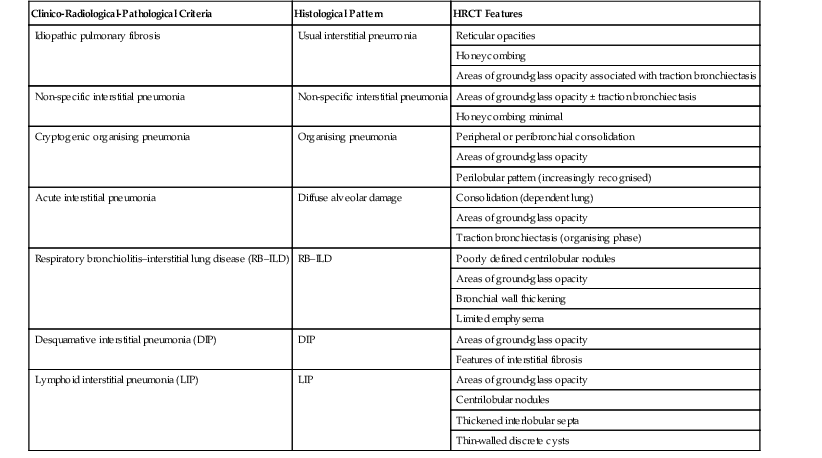
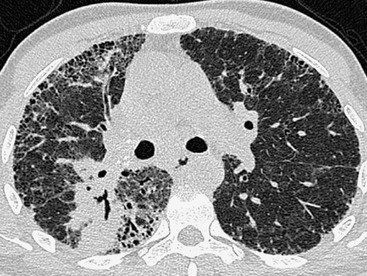
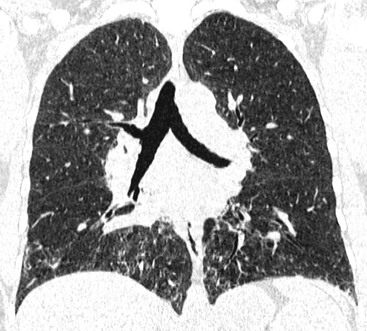
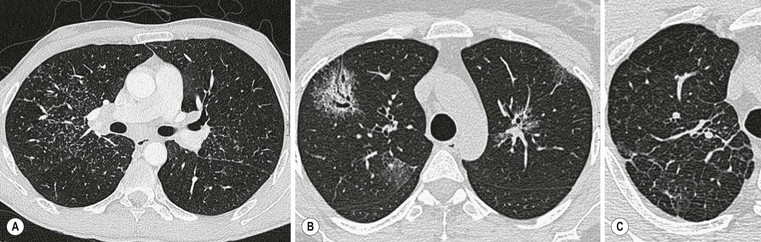
The term idiopathic pulmonary fibrosis (IPF) is applied to patients with a histologic and/or computed tomography (CT) pattern of usual interstitial pneumonia (UIP) and compatible clinical and imaging features.12 Other causes of a UIP-type pattern on histology include chronic hypersensitivity pneumonitis, asbestosis, connective tissue disease and rarely drugs. The pathological features of UIP are the presence of fibroblastic foci, normal areas, dense fibrosis and honeycombing; the crucial finding is of areas of fibrosis at different stages of maturity. The characteristic and virtually pathognomonic appearance of UIP on HRCT is of a predominantly subpleural bibasal reticular pattern within which there are areas of honeycomb destruction. As the disease progresses, it often appears to ‘creep’ around the periphery of the lung to involve the anterior aspects of the upper lobes (Fig. 16-1). The presence of ground-glass opacification is not a dominant feature and, when present, there is usually obvious traction bronchiectasis and bronchiolectasis.13 Mediastinal lymphadenopathy (up to approximately 2.5 cm) unrelated to infection or malignancy is a frequent accompaniment.14
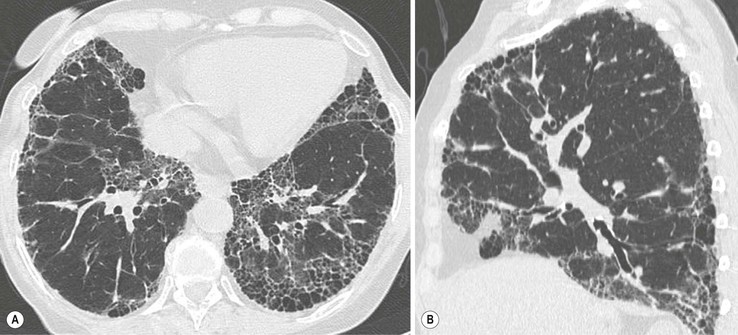
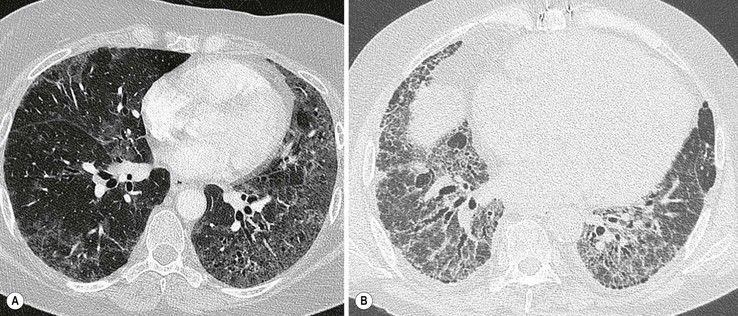
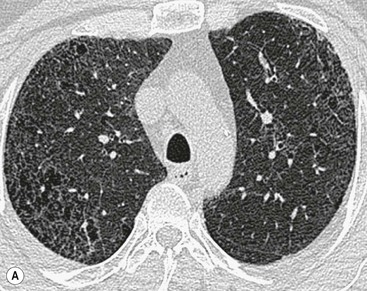
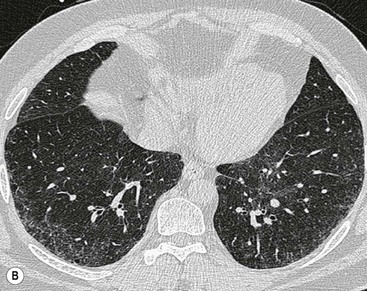
In the appropriate clinical setting, the presence of a classical UIP pattern on HRCT is sufficient for the diagnosis of IPF, without the need for surgical biopsy. Therefore, the primary role of HRCT is to separate patients with UIP from those with non-UIP pattern as they may have a substantially different prognosis. A confident HRCT diagnosis of UIP is not usually made unless honeycombing is present. If honeycombing is absent, but the imaging features otherwise meet criteria for UIP (especially when the pattern is characterised by reticular opacities in predominantly peripheral and basal distribution), the imaging features are regarded as representing possible UIP, and surgical lung biopsy is necessary to make a definitive diagnosis.12 However, in patients whose HRCT does not demonstrate either a classical or a possible UIP pattern, the surgical lung biopsy may still demonstrate UIP pattern on histopathology. The majority of these atypical UIP cases are usually characterised by predominant ground-glass opacity. Thus, their HRCT appearances frequently mimic non-specific interstitial pneumonia (NSIP) (Fig. 16-2).15,16 HRCT has also a role in predicting survival. A study by Flaherty et al. suggested that patients with histological UIP who had definite UIP by HRCT criteria had a worse prognosis than those who had interdeterminate (or atypical) HRCT findings.15 The extent of fibrosis, traction bronchiectasis and honeycombing on HRCT are also predictive of both survival and mortality in IPF.13,17
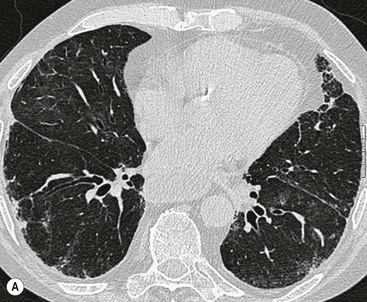
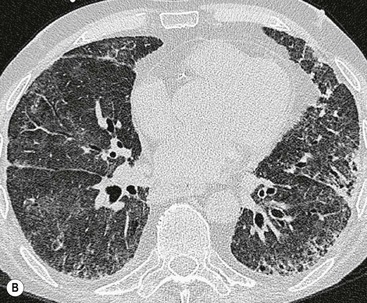
The rapid development of a diffuse increase in the attenuation of lung parenchyma in patients with IPF should raise the possibility of an accelerated phase (also known as an acute exacerbation) of the disease (Fig. 16-2), concurrent pulmonary oedema, or rarely an atypical infection.12,18 Other complications include lung cancer and pulmonary tuberculosis (Fig. 16-3); the latter usually has atypical appearances on CT caused by the presence of underlying lung fibrosis.19,20
Non-Specific Interstitial Pneumonia
Non-specific interstitial pneumonia (NSIP) is characterised by varying degrees of interstitial inflammation and fibrosis without the specific features that allow a diagnosis of UIP.21 While NSIP may have significant fibrosis, it is usually temporally uniform (in comparison to UIP), and fibroblastic foci and honeycombing are absent or scanty. Although the clinical features of idiopathic NSIP resemble those of UIP, prognosis is considerably better. Non-idiopathic NSIP is most often found on lung biopsy in patients with connective tissue disease and may be the predominant histopathological pattern in some cases of drug-induced lung disease and chronic hypersensitivity pneumonitis. On HRCT, ground glass with or without associated distortion of airways is usually the dominant pattern (Fig. 16-4). Reticular abnormalities are common, but honeycombing is sparse or absent even when other signs of fibrotic changes are evident. Abnormalities are usually peribronchovascular or peripheral, although they may sometimes spare the subpleural lung.21 In general, NSIP may be distinguished from UIP on CT by a more prominent component of ground-glass attenuation and a finer reticular pattern in the absence of honeycombing.22 However, the variability of CT appearances reflects the heterogeneity of the pathological processes encompassed by NSIP and a confident diagnosis of NSIP based on CT alone is less readily made than in cases of UIP. Consolidation is reportedly a highly variable feature (0–98%) and this discrepancy probably reflects the fact that some patients with non-idiopathic NSIP have significant amounts of histological organising pneumonia, making classification of individual cases difficult.23
Cryptogenic Organising Pneumonia
This is considered in the section on airspace disease.
Respiratory Bronchiolitis–Interstitial Lung Disease and Desquamative Interstitial Pneumonia
These two entities are considered together because of their strong association with cigarette smoking. All cigarette smokers have, to some degree, inflammation around their small airways (‘respiratory bronchiolitis’) but this is clinically unimportant and not considered further here. Patients with respiratory bronchiolitis–interstitial lung disease generally present with an insiduous onset of dyspnoea and cough. The chest radiograph is relatively insensitive for the detection of RB–ILD and desquamative interstitial pneumonia (DIP) and a normal chest radiograph has been reported in up to 20% of patients with RB–ILD and 25% in DIP.24,25
On HRCT, the features of RB–ILD include areas of patchy ground-glass opacification (resulting from macrophage accumulation within alveolar spaces and alveolar ducts) and poorly defined low-attenuation centrilobular nodules (Fig. 16-5). In addition, upper lobe centrilobular emphysema, usually of very limited extent, and areas of air trapping may be present, the latter reflecting the bronchiolitic element of this entity.26 Some patients may show scattered thickening of the interlobular septa and features of interstitial fibrosis, but this is not the dominant pattern.27
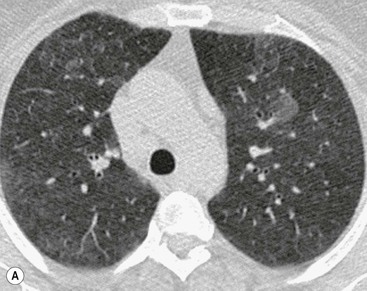
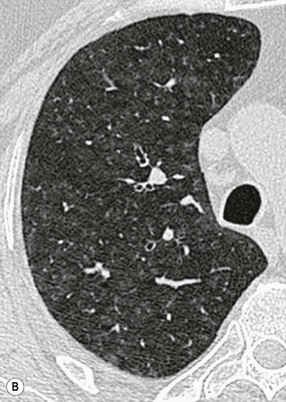
Ground-glass opacification is the dominant feature seen in DIP (Fig. 16-6). The distribution is typically lower zone, peripheral and may be patchy or geographic.28 In some patients there are HRCT features of established fibrosis (in the form of architectural distortion with dilatation of some bronchi), usually of limited extent. The majority of patients with DIP or RB–ILD have a relatively stable clinical course. Smoking cessation is an important part of the management of patients, but the influence of smoking on the clinical course of these patients has not been fully delineated; some patients have persistent abnormalities on HRCT even with smoking cessation and corticosteroid therapy. Because of the significant overlap between the clinical, imaging and histological features of DIP and RB–ILD and to a lesser extent between these two patterns and Langerhans cell histiocytosis (LCH) and interstitial fibrosis, the global term ‘smoking related-interstitial lung disease’ (SR-ILD) has been proposed to encompass DIP, RB–ILD, LCH and interstitial fibrosis (Fig. 16-7).27,29
Acute Interstitial Pneumonia/Diffuse Alveolar Damage
Acute interstitial pneumonia can be regarded as an idiopathic form of ARDS and is histologically (and clinically) distinct from the other interstitial pneumonias.9 The histological pattern seen in AIP is that of diffuse alveolar damage (DAD), which is also found in infection, connective tissue disease, drug toxicity and toxic fume inhalation. DAD has an acute exudative phase and a subsequent organising and fibrotic phase. Lung biopsy shows diffuse involvement with temporal homogeneity, which implies lung injury due to a single event. The chest radiograph shows bilateral patchy airspace opacification.30 HRCT demonstrates a combination of ground-glass opacification, consolidation, bronchial dilatation and architectural distortion.31 Ground-glass opacification on HRCT is found in all three phases of AIP, but coexistent traction bronchiectasis probably reflects the early incorporation of established fibrosis.32 Follow-up CT shows reticular opacities consistent with residual fibrosis. Anterior non-dependent fibrotic damage in survivors secondary to barotrauma has also been reported.33
Lymphoid Interstitial Pneumonia
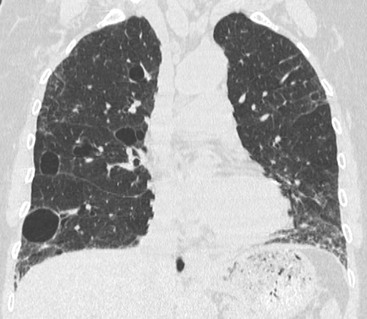
The term ‘lymphoid interstitial pneumonia’ (LIP) was proposed by Liebow and Carrington to describe a disease entity characterised by a widespread interstitial lymphoid infiltrate of the lung, resembling lymphoma but with a clinical course more akin to a chronic interstitial pneumonia. Although in the past LIP has been considered by some to be a pulmonary lymphoproliferative disorder, evolution to frank lymphoproliferative disease is rare and thus LIP remains within the group of interstitial pneumonias.9,34 Classically, LIP occurs in association with autoimmune diseases, most often Sjögren’s syndrome. Other diseases associated with LIP include dysproteinaemias, autologous bone marrow transplantation and viral, mycobacterial and human immunodeficiency virus (HIV) infections.34 LIP is approximately twice as frequent in women and symptoms of progressive cough and dyspnoea usually predominate. Common HRCT findings are nodules of varying sizes (which may be ill-defined), areas of ground-glass opacification, thickened bronchovascular bundles, interlobular septal thickening and thin-walled cysts (1–30 mm) (Fig. 16-8).35 Airspace disease, large nodules and pleural effusions are rare in these patients. The cysts in LIP are usually discrete, sometimes clustered and tend not to be subpleural.36
Idiopathic Pleuroparenchymal Fibroelastosis
Little is known regarding aetiology of idiopathic pleuroparenchymal fibroelastosis, but recurrent infections are a feature in some patients and a few cases have been reported in association with previous bone marrow or lung transplantation.37 IPPFE is characterised by dense established intra-alveolar fibrosis containing prominent elastosis, and dense fibrous thickening of the visceral pleura; these changes have a striking upper zone predominance.10,11 HRCT appearances consist of irregular pleural thickening and ‘tags’ in the upper zones that merge with fibrotic changes in the subjacent lung (Fig. 16-9; Table 16-3).10
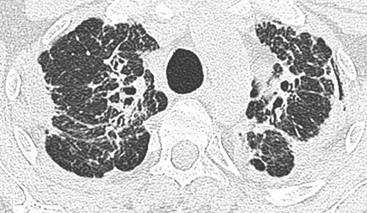
TABLE 16-3
Causes of Bilateral Upper Lobe Fibrosis

Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder of unknown aetiology. As a consequence, the diagnosis of this syndrome is defined by the presence of characteristic clinical and radiological data along with histological evidence of non-caseating granuloma. Granulomas in the lung have a characteristic distribution along the lymphatics in the bronchovascular sheath and, to a lesser extent, in the interlobular septa and subpleural lung regions. Sarcoidosis is a disease of young adults, with a peak incidence in the second to fourth decades. The hilar and mediastinal nodes and the lungs are affected clinically much more commonly than any other organ or system. They are followed in decreasing order of frequency by the skin (26%), peripheral lymph nodes (22%), eyes (15%), spleen (6%), central nervous system (4%), parotid glands (4%) and bones (3%).38
Pulmonary involvement accounts for most of the morbidity and mortality associated with sarcoidosis. Sarcoidosis is traditionally staged according to its appearance on the chest radiograph: stage I, lymphadenopathy; stage II, lymphadenopathy with parenchymal opacity; stage III, parenchymal opacity alone.39 Low stages at presentation are reported to have a better prognosis than high stages, although the precision and clinical usefulness of such ‘staging’ is questionable.
Lymphadenopathy
Sarcoidosis is characterised by bilateral, symmetrical hilar and paratracheal lymphadenopathy. Some degree of lymphadenopathy is evident on a chest radiograph in about 70–80% of patients at some time during the course of the condition. Hilar lymph node enlargement ranges from the barely detectable to the massive and gives the hila a lobulated and usually well-demarcated outline. Occasionally hilar lymphadenopathy appears to be asymmetrical or, in 1–5% of cases, may even be strictly unilateral although this is distinctly unusual.40,41
Paratracheal lymphadenopathy may be bilateral or unilateral and in the latter instance is usually right-sided. The most common manifestation of left-sided lymphadenopathy is enlargement of the aortopulmonary window nodes—a common and characteristic feature on the chest radiograph. Other mediastinal nodes (anterior prevascular, posterior and subcarinal) are often not identified as being enlarged on the chest radiograph but on CT are seen to be affected in about half of patients.42 In 90% of patients with lymphadenopathy, nodal enlargement is maximal on the first radiograph and usually disappears within 6–12 months. Recurrence of lymphadenopathy is exceedingly rare.43
The affected lymph nodes may calcify, sometimes in a characteristic eggshell fashion. This latter feature is shared by only a few conditions such as silicosis and histoplasmosis. The calcification is of variable intensity but may be relatively light, and the affected lymph nodes are usually small in volume and evenly distributed throughout the mediastinum and hila (very different from calcified nodes due to tuberculous infection which usually follow a drainage path).44 About 40% of patients presenting with nodal enlargement will develop parenchymal opacities, usually within a year, and of these about one-third will go on to have persistent (fibrotic) shadowing. Nodal enlargement does not develop after parenchymal opacities have appeared.
Parenchymal Changes
Parenchymal changes probably occur histologically in the majority of patients but are only detected on the chest radiograph in 50–70% of cases. Characteristically, parenchymal abnormalities appear as the nodal enlargement is subsiding (in lymphoma such abnormalities tend to progress in unison). The most common radiographic pattern, seen in 75–90% of patients with parenchymal opacities, is of rounded or irregular nodules 2–4 mm in diameter, which are usually well defined. Smaller or larger opacities are not uncommon, though they rarely exceed 5 mm. All zones can be affected but there is usually a mid and upper zonal predominance. The second most common pattern, seen in 10–20% of patients with parenchymal opacity, is patchy airspace consolidation. Opacities sometimes contain air bronchograms and have ill-defined margins that commonly break up into a nodular pattern. They tend to involve predominantly the peribronchovascular regions of the middle and upper lung zones, although they may be diffuse or, occasionally, have a subpleural predominance.
The parenchymal opacities described above will clear completely in about two-thirds of cases and progress to fibrosis in one-third. Permanent fibrotic shadowing is unusually coarse, with a mid and upper zone predominance. The radiographic pattern consists of coarse linear opacities with evidence of volume loss and ring shadowing caused by bullae or traction bronchiectasis. Occasionally a conglomerate opacity develops, resembling progressive massive fibrosis. Pulmonary hypertension, bullous disease with or without mycetoma formation, and pneumothorax are all recognised complications of this fibrotic stage.
High-Resolution Computed Tomography Features
Despite the better delineation of parenchymal disease on HRCT, it is not recommended as part of the initial diagnostic work-up in patients with suspected sarcoidosis; its greatest use is in patients who present with an atypical chest radiograph.45 Parenchymal opacities are well demonstrated on HRCT (Fig. 16-10) and HRCT appearances have a high sensitivity and specificity for the diagnosis of sarcoidosis.45,46 The most consistent pulmonary parenchymal abnormality is the presence of nodular opacities (1–5 mm) distributed in a perilymphatic fashion, predominantly along the bronchovascular bundles and subpleurally and, to a lesser extent, along interlobular septa.46 Other findings include irregular and beaded interfaces, larger ill-defined nodules with/without an air bronchogram, patchy ground-glass opacities and occasional interlobular septal thickening. In advanced disease there is evidence of fibrosis, predominantly in the perihilar regions of the middle and upper lung zones.47 Air trapping is a common HRCT feature of sarcoidosis and its presence shows a good correlation with indices of small airways disease on pulmonary function tests.48 Less common parenchymal changes seen in sarcoidosis evolve if the small nodules are confluent to form larger nodules (nodular sarcoidosis, galaxy sign) or if the nodules are so small, being below the resolution of the HRCT, that they appear as areas of ground glass (alveolar sarcoidosis).
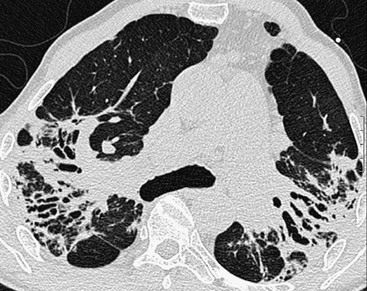
Previous HRCT studies have shown that areas of parenchymal consolidation and ground-glass opacity are usually reversible, whereas resolution is not expected in patients showing reticulation and architectural distortion.49,50 Classic fibrotic changes include linear opacities (radiating laterally from the hilum), fissural displacement, bronchovascular distortion (bronchiectasis) and honeycombing concentrated in the upper zones (Fig. 16-11).51
Despite this distinction between reversible and irreversible disease on HRCT, studies comparing HRCT assessment of disease activity to clinical, scintigraphic and bronchoscopic findings have yielded contradictory results.52 Hence, HRCT is not generally used to guide prognosis in patients with sarcoidosis.
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis (EAA), is an immunologically mediated lung disease characterised by an inflammatory reaction to specific antigens contained in a variety of organic dusts. Common causes include avian proteins (e.g. bird breeder’s lung) and thermophilic bacteria present in mouldy hay (farmer’s lung), mouldy grain (grain handler’s lung), or heated water reservoirs (humidifier or air conditioner lung).53 These antigens reach the alveoli where they provoke an immunological reaction that includes both type III (immune complex response) and type IV (cell-mediated) mechanisms. The cell-mediated response results in a delayed hypersensitivity reaction and the presence of granulomatous inflammation within the pulmonary interstitium. Interestingly, several studies have shown that cigarette smoking has a suppressive effect that interferes with the immunopathological process that ultimately leads to HP.54
The clinical features of hypersensitivity pneumonitis are characteristic. Approximately 6 h after exposure the patient develops fever, chills, dyspnoea and cough. There is no eosinophilia, and wheeze is not a prominent feature. The radiological findings are influenced by the stage of the disease. Typical HRCT findings include centrilobular nodules, which are typically poorly defined, < 5 mm in diameter, centrilobular and seen throughout the lung, although a mid to lower lung zone predominance has been variably reported (Fig. 16-12).53,55 Ground-glass opacity is most common in the acute phase but may also be a feature of subacute and chronic HP, especially if there is ongoing exposure.56 A mosaic attenuation pattern is common in HP; the presence of lobular areas of decreased vascularity that show air trapping on expiratory HRCT reflect the coexisting bronchiolitis caused by antigen deposition in the small airways (Fig. 16-13).57 The combination on HRCT of features of infiltrative (ill-defined nodules and ground-glass opacity) and small airways disease may be remarkably similar to that seen in patients with RB–ILD; however, the distinction can usually be made with knowledge of the smoking history. The presence of thin-walled lung cysts is also an occasional feature in subacute HP. Cysts range in size from 3 to 25 mm and resemble those seen in lymphoid interstitial pneumonia, although their pathogenesis remains uncertain.58 Emphysema is a reported sequela of farmer’s lung and a study has demonstrated that in hypersensitivity caused by farmer’s lung, emphysema was a more prominent feature than honeycombing/fibrosis (even in never smokers) and was seen in approximately one-third of patients.59
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


