Hirayama disease (juvenile muscular atrophy of distal upper extremity) is a cervical myelopathy. Predominantly affecting male adolescents, it is characterized by progressive muscular weakness and atrophy of distal upper limbs, followed by spontaneous arrest within several years. Although the cause of cervical myelopathy remains unclear, neuropathologic and neuroradiologic findings suggest a forward displacement of the posterior cervical dural sac during neck flexion, causing compression of the cervical cord, and results in atrophic and ischemic changes in the anterior horn. A good understanding of Hirayama disease is essential because early recognition and management can effectively halt the progressive deterioration.
Hirayama disease is a benign, self-limiting cervical myelopathy first brought to attention by Hirayama in 1959. During the past half century, researches have established this disease as a new entity that differs from motor neuron disease because of spinal cord compression by the posterior dural sac during neck flexion. Primarily affecting male adolescents, it is clinically characterized by a progressive course of distal upper-limb muscular weakness and atrophy, followed by a spontaneous arrest within several years. Early recognition of the disease allows early intervention, which has been shown to stop disease progression. This article reviews the clinical presentation, pathophysiology, image findings, and treatment of Hirayama disease, because a thorough understanding of the disease is essential for early recognition and treatment.
In 1959, Hirayama and colleagues published 12 cases distinguishing a new clinical entity from what was originally thought to be a type of progressive and degenerative motor neuron disease. The term “juvenile muscular atrophy of unilateral upper extremity” was applied. This new clinical entity was further consolidated by following reports of 20 patients in 1963 and 38 patients in 1972. The patients are generally in their teens and early twenties, predominantly male, with insidious onset of weakness and muscle wasting involving unilateral distal upper limb, followed by variable period of progression and spontaneous arrest within several years.
Since then, there have been many reports of Hirayama disease, mostly from Japan and other Asian countries, as well as few from other countries. However, early reports in the 1960s and 1970s emphasized clinical features and electrophysiological evaluations. It was not until 1982 that the first autopsy case was obtained by Hirayama and colleagues, which revealed anterior-posterior flattening of the lower cervical cord associated with ischemic and atrophic changes of the anterior horn cells. Pathologic evidence of focal ischemic cervical poliomyelopathy of the disease prompted neuroradiologic investigation in search of the anatomic pathophysiology of the spinal cord and canal. Earlier studies were conducted using myelography and CT myelograpy. Since MR imaging equipment became available and widely accessible in the late 1980s, several publications have provided valuable findings in neck-flexion and neutral-position MR imaging of the cervical spine that have assisted the diagnosis of this disease. Imaging findings revealed dynamic changes of the cervical spine, spinal canal, and spinal cord during neck flexion, mainly in the forward displacement of posterior dural sac, causing anterior-posterior flattening and asymmetric atrophy of the lower cervical cord, which corresponded with the pathologic findings.
Several names have been used in publications to describe this disease, including benign juvenile brachial spinal muscular atrophy, juvenile asymmetric segmental spinal muscular atrophy, juvenile muscular atrophy of the distal upper extremity, monomelic amyotrophy, and oblique amyotrophy. Hirayama disease is still the most commonly used named throughout publications in neurology and neuroradiology, as well as related fields.
Demographics
Hirayama disease occurs mainly in people in their teens and early twenties with a significant male predominance. The mean age of onset ranges from 15 to 20 years of age in different studies. The degree of male preponderance reported in large series ranged from purely male to a male/female ratio of 2.8:1. A delay of age of onset after the peak age of normal growth curve was observed in several studies. Hirayama observed that the peak of onset age is approximately 2 years later than the peak of longitudinal growth curve of juveniles in Japan. Based on this finding, Hirayama speculated that disproportional growth between the vertebral column and the contents of the spinal canal, especially the dural sac, during the juvenile growth spurt underlies this phenomenon. Tashiro and colleagues suggested that the difference in the male and female incidence of the disease could be explained by a more rapid increase in height of males at puberty compared with females.
Hirayama disease is nonfamilial in most patients. There are, however, a few reports of familial cases. Nine familial cases have been reported in the English literature. So far, there are no published reports regarding genetics information that maybe related to familial Hirayama disease. The explanation for the familial occurrence is indeed interesting and needs further studies.
As Hirayama disease became more recognized, several cases and large series have accumulated. There are an exceedingly large number of cases in Japan and more cases reported from other Asian countries in the past 3 decades, particularly in China, Taiwan, Malaysia, India, and Sri Lanka, but relatively fewer cases from Europe and North America. Whether there might be an ethnic factor in this disorder is still unclear.
Clinical features of Hirayama disease
Patients with Hirayama disease present with insidious onset and slow progression of muscle weakness and muscular atrophy of the distal upper limb, including thenar, hypothenar, interossei muscles, and wrist flexors and extensors, with sparing of brachioradialis muscles ( Fig. 1 ). The border of muscular atrophy runs obliquely over the volar and dorsal surfaces of the forearm, therefore it is also referred to as oblique amyotrophy. Both extensor and flexor muscles of the fingers and wrists are involved, particularly the finger extensors and wrist flexors. Some investigators reported asymmetric atrophy was more severe on the ulnar than the radial side in the flexor and extensor compartments. The phenomenon of cold paresis is widely observed in the patients, characterized by exacerbating of finger weakness on exposure to cold environment. Kijima and colleagues proposed that this is caused by conduction block of the muscle fiber membrane in reinnervated muscles after active denervation. When cold paresis is induced, compound muscle action potentials show delayed latencies and decreased amplitude after high-frequency repetitive nerve stimulation. Resting fasciculation is not observed, but contraction fasciculation is well documented. It is characterized by fascicular twitching in the extensor side of forearm muscles or tremor-like movement of the fingers during stretching. Fatigue is the most common symptom (33%), followed by cold paresis, atrophy, and tremor. Although most patients reported absence of subjective or objective sensory disturbances, a few patients show slight hypoesthesia in a localized area of the hand. The muscle stretch reflexes of the arms and legs are within normal range. There are no accompanying neurologic symptoms such as cranial nerve involvement, pyramidal signs, sensory disturbance, and urinary disturbance. Muscle biopsy and needle electromyography show denervation of the atrophied muscles. Nerve conduction velocity and analysis of the cerebrospinal fluid are almost normal.
Patients with Hirayama disease commonly show initial unilateral monomelic atrophy, but it is asymmetrically bilateral in some and, occasionally, symmetric. The right upper limb is more frequently involved in Hirayama disease, regardless of the handedness. Huang and colleagues reported more conspicuous manifestation in right upper limb with a right/left ratio of 3:1. Often, the disease progressed to involve the other side in an asymmetric manner. A bilateral symmetric form of Hirayama disease has occasionally been reported. The incidence was 3.1% by a National Japan Survey and 10% by Pradhan, who reported 11 cases of bilateral involvement in a series of 106 patients. He proposed that the “bilateral symmetric Hirayama disease” is a more severe variant of Hirayama disease based on more severe and significant clinical and MR imaging findings. In his series, patients with bilateral involvement showed wider range of involvement, including partial involvement of brachioradialis and biceps brachii. The patients had significant weakness of the forearm and small muscles of the hands; required assistance in daily activities, such as buttoning clothes, holding spoons, and combing hair; had autonomic dysfunction, such as excessive sweating in the hands and cold extremities; and had occasional brisk deep tendon reflexes in the lower limbs. In patients with bilateral involvement, two patterns of progression from one upper limb to the other were observed. The first was that muscle symptoms developed in the other arm during progression of the initial involved arm (with about a 1-year delay in onset). The second was that muscle symptoms developed approximately 2.5 years after cessation of progression in the first arm.
The disease follows a progressive course for a variation of period, with spontaneous arrest within several years. Symptoms generally progress for 2 to 4 years after onset. The duration of progression varies widely, ranging from 16.8 to 73.2 monthes. Progression of the disease ceased within 5 years in most patients (around 90%). Early arrest of the progression is essential for any possibility of improvement.
Clinical features of Hirayama disease
Patients with Hirayama disease present with insidious onset and slow progression of muscle weakness and muscular atrophy of the distal upper limb, including thenar, hypothenar, interossei muscles, and wrist flexors and extensors, with sparing of brachioradialis muscles ( Fig. 1 ). The border of muscular atrophy runs obliquely over the volar and dorsal surfaces of the forearm, therefore it is also referred to as oblique amyotrophy. Both extensor and flexor muscles of the fingers and wrists are involved, particularly the finger extensors and wrist flexors. Some investigators reported asymmetric atrophy was more severe on the ulnar than the radial side in the flexor and extensor compartments. The phenomenon of cold paresis is widely observed in the patients, characterized by exacerbating of finger weakness on exposure to cold environment. Kijima and colleagues proposed that this is caused by conduction block of the muscle fiber membrane in reinnervated muscles after active denervation. When cold paresis is induced, compound muscle action potentials show delayed latencies and decreased amplitude after high-frequency repetitive nerve stimulation. Resting fasciculation is not observed, but contraction fasciculation is well documented. It is characterized by fascicular twitching in the extensor side of forearm muscles or tremor-like movement of the fingers during stretching. Fatigue is the most common symptom (33%), followed by cold paresis, atrophy, and tremor. Although most patients reported absence of subjective or objective sensory disturbances, a few patients show slight hypoesthesia in a localized area of the hand. The muscle stretch reflexes of the arms and legs are within normal range. There are no accompanying neurologic symptoms such as cranial nerve involvement, pyramidal signs, sensory disturbance, and urinary disturbance. Muscle biopsy and needle electromyography show denervation of the atrophied muscles. Nerve conduction velocity and analysis of the cerebrospinal fluid are almost normal.
Patients with Hirayama disease commonly show initial unilateral monomelic atrophy, but it is asymmetrically bilateral in some and, occasionally, symmetric. The right upper limb is more frequently involved in Hirayama disease, regardless of the handedness. Huang and colleagues reported more conspicuous manifestation in right upper limb with a right/left ratio of 3:1. Often, the disease progressed to involve the other side in an asymmetric manner. A bilateral symmetric form of Hirayama disease has occasionally been reported. The incidence was 3.1% by a National Japan Survey and 10% by Pradhan, who reported 11 cases of bilateral involvement in a series of 106 patients. He proposed that the “bilateral symmetric Hirayama disease” is a more severe variant of Hirayama disease based on more severe and significant clinical and MR imaging findings. In his series, patients with bilateral involvement showed wider range of involvement, including partial involvement of brachioradialis and biceps brachii. The patients had significant weakness of the forearm and small muscles of the hands; required assistance in daily activities, such as buttoning clothes, holding spoons, and combing hair; had autonomic dysfunction, such as excessive sweating in the hands and cold extremities; and had occasional brisk deep tendon reflexes in the lower limbs. In patients with bilateral involvement, two patterns of progression from one upper limb to the other were observed. The first was that muscle symptoms developed in the other arm during progression of the initial involved arm (with about a 1-year delay in onset). The second was that muscle symptoms developed approximately 2.5 years after cessation of progression in the first arm.
The disease follows a progressive course for a variation of period, with spontaneous arrest within several years. Symptoms generally progress for 2 to 4 years after onset. The duration of progression varies widely, ranging from 16.8 to 73.2 monthes. Progression of the disease ceased within 5 years in most patients (around 90%). Early arrest of the progression is essential for any possibility of improvement.
Pathophysiology
Pathologic study of this disease was not available until 1982 owing to its benign course. Before neuropathology, there were strenuous debates about whether the disease could be classified as a new disease entity. Some investigators considered the illness to be a variant of degenerative motor neuron disease, traumatic injury of the cervical spinal cord, prolonged survival of acute anterior poliomyelitis, or syringomyelia localized in the anterior horn, in addition to other diseases.
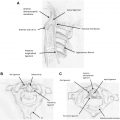
The autopsy case was a 38 year-old patient who experienced left distal upper limb muscular atrophy and weakness associated with cold paresis and fine tremor starting at the age of 15. The symptoms progressed for approximately 1 year before spontaneous arrest. Thereafter, the symptom remained unchanged until the age of 38 when he was diagnosed with lung cancer and died 3 months later. Gross pathology showed evident anterior-posterior flattening of the cervical cord, particularly at C7 and C8. Microscopically, bilateral anterior horns were reduced to less than half of the normal anterior-posterior diameter, most severely at C7 and C8, and predominantly on the left side, corresponding with the patient’s clinical symptoms ( Fig. 2 ). The anterior horn lesions were characterized by central necrosis without cavity formation, decrease in the number of large and small nerve cells in the periphery with degenerative changes, and mild astrogliosis without macrophage infiltration. There were no abnormal vascular proliferations or amyloid deposition. The posterior horns, posterior roots, white matter, and the intra medullary and extramedullary vessels were normal. There were no metastatic or spondylolytic changes of the cervical vertebrae and cord. Loss of large and small neurons, a weak gliosis, and central necrosis of the anterior horn were not supportive of degenerative motor neuron disease or other diseases proposed before neuropathology. Hirayama concluded that the changes could be ischemic in origin.
In combination with clinical symptoms, the neuropathological finding of ischemic change of the anterior horn resembles another ischemic neuronal disease, téphromalacie antérieure (anterior tephromalacia). Reported by Marie and Foix in 1912, téphromalacie antérieure consist of anterior horn infarction of lower cervical cord from occlusion of the spinal arteritis due to syphilitic arteritis or arteriosclerosis. Clinically, téphromalacie antérieure also shows muscular wasting restricted to the distal upper limbs, but in middle-aged or elderly people. In 1969, after a study of spinal cord of the aged, Lapresle reported that the anterior horn, particularly the neurons in its central portion, was the most vulnerable to ischemia. Although the mechanisms of cervical cord anterior horn damage may be different, both clinical and pathologic findings suggested a similar pathogenesis between the two diseases owing to circulatory insufficiency of the anterior horns of lower cervical cord. The name focal ischemic cervical poliomyelopathy was given for diseases with similar pathologic finding of anterior horn ischemia.
After the publication of the first autopsy case of Hirayama disease, the neuropathological findings of ischemic cervical poliomyelopathy encouraged neuroradiologic investigations of the interior changes in the spinal canal. Imaging studies revealed dynamic changes of the spinal cord and dural sac during anterior flexion of the neck in patients with Hirayama disease ( Fig. 3 ). These findings include overstretch of the cord, forward displacement of the posterior dural sac, and asymmetric compression and anterior-posterior flattening of the lower cervical cord (see later discussion on more detailed image findings). The dural displacement decreases gradually with increasing age, which also corresponds well with the duration and clinical course of the disease. These dynamic changes are unequivocal findings confined to early, progressive stage of the disease. The absence of forward displacement of the dural sac and cord compression in elderly patients whose disease had arrested suggested that this dynamic compression has a pathogenetic significance.