Vascular pathologies of brain comprise a heterogeneous group of abnormalities such as vasculitis, dural arteriovenous malformations, carotid-cavernous fistulas, cerebral venous thrombosis, and intracerebral hemorrhage. Modern imaging techniques are increasingly unraveling these vascular lesions of the brain at a stage when many of them are still asymptomatic. This article focuses on acquired vascular pathologies of brain prevalent in tropical countries that are encountered in clinical practice.
Vascular pathologies of brain comprise a heterogeneous group of abnormalities such as vasculitis, dural arteriovenous malformations, carotid-cavernous fistulas, cerebral venous thrombosis (CVT), and intracerebral hemorrhage (ICH). Modern imaging techniques are increasingly unraveling these vascular lesions of the brain at a stage when many of them are still asymptomatic. This article focuses on acquired vascular pathologies prevalent in the tropical countries. Some of the common vascular pathologies of brain lesions encountered in clinical practice are enumerated here ( Box 1 ).
- •
Vasculitis and Vasculopathy
- ○
Primary vasculitis
- ▪
Primary central nervous system angiitis
- ▪
Giant cell arteritis
- ▪
- ○
Secondary Vasculitis
- ▪
Systemic vessel wall disease
- •
Takayasu arteritis
- •
Moyamoya disease
- •
Polyarteritis nodosa
- •
- ▪
Autoimmune Vasculitis
- ▪
Infectious Vasculitis
- •
Bacterial infection—acute septic meningitis, tubercular meningitis
- •
Fungal infection— Candida , aspergillosis, coccidiomycosis, mucormycosis
- •
Viral infection—varicella zoster infection, human immunodeficiency virus vasculopathy
- •
Parasitic infection—neurocysticercosis
- •
Mycotic aneurysms
- •
- ▪
Drug-induced vasculitis
- ▪
Radiation vasculopathy
- ▪
- ○
Reversible cerebral vasoconstriction syndrome
- ○
Arterial dissections
- ○
- •
Dural Arteriovenous Malformations
- •
Cerebral Venous Thrombosis
- •
Nontraumatic hemorrhage
- ○
Intracerebral hemorrhage
- ○
Subarachnoid hemorrhage
- ○
- •
Caroticocavernous Fistula
- •
Posterior reversible Encephalopathy Syndrome
Vasculitis and vasculopathy
Cerebral vasculitis is heterogeneous group of multisystem disorders characterized pathologically by inflammation with or without necrosis of the blood vessel wall. Vasculitis, like inflammation in other tissues, may be caused by many different agents and pathogenic mechanisms; however, these different causes produce only a limited number of histologic expressions of injury—ischemia following vessel lumen compromise and hemorrhage following vessel wall disruption. Therefore, the same clinical manifestations can result from etiologically and pathogenetically different vasculitic diseases.
Important clinical factors that merit consideration include age, sex, and ethnic origin of the patient, presence of skin lesions (ulcers, palpable purpura), size of vessel involvement (small, medium, and large), involvement of other organs (in particular kidneys, lungs, and paranasal sinuses), use of medications, drug abuse, and neurologic signs including cognitive deterioration, focal deficit, transient ischemic attack, stroke, and thunder-clap headache (TCH). Relevant laboratory tests should include erythrocyte sedimentation rate (ESR), rheumatoid factor, C-reactive protein, complement level, immune status and antibodies in blood and cerebrospinal fluid (CSF), protein and cell count in CSF, and assessment of coagulation factors. Chest radiographs may reveal asymptomatic pulmonary involvement.
Computed tomography (CT) of the brain is less useful for the diagnosis of acute central nervous system (CNS) vasculitis because of limited spatial resolution, and thus can demonstrate only large ischemic infarctions well, although it can show parenchymal brain calcifications found within old ischemic lesions. Magnetic resonance (MR) imaging plays a crucial role in the workup of patients of suspected vasculitis, even though the abnormalities found on MR imaging are not diagnostic. Involvement of small perforating arteries results in ischemic lesions localized in the deep or subcortical white matter and the basal ganglia. When larger arteries are occluded, the resulting infarctions are found in the cortical gray and white matter. During the acute stage of the infarction, diffusion-weighted MR imaging can distinguish acute from chronic ischemic abnormalities. Within a few days, acute infarctions progress to a subacute stage with neoangiogenesis, and may enhance following contrast administration. In addition, wall thickening and intramural contrast uptake are frequent findings in patients with active cerebral vasculitis affecting large brain arteries.
Important clinical features and literature on different forms of vasculitis are briefly reviewed here.
Primary Vasculitis
Primary CNS angiitis (granulomatous angiitis)
Primary angiitis of the central nervous system (PACNS), also called primary CNS vasculitis, is an idiopathic inflammatory condition affecting medium-sized to small arteries, arterioles, and venules of brain parenchyma and meninges. Rarely, PACNS affects the spinal cord. PACNS is most often observed in the fifth to sixth decade; however, several pediatric cases have also been reported. Acute or subacute onset confusion, headache, change of personality, paresis, cranial neuropathy, hallucinations, or loss of consciousness are often presenting signs but, as such, nonspecific. Laboratory findings include elevated inflammatory markers, particularly the ESR, and CSF analysis may reveal elevated opening pressure, raised protein levels, and/or lymphocytic pleocytosis.
Pathologically, transmural involvement of the vessel wall is commonly seen with dense infiltration of inflammatory cells, primarily lymphocytes and large mononuclear cells, with or without the presence of granulomas. There may be fibrinoid necrosis in the vessel wall, and inflammation in the leptomeningeal and/or parenchymal vessels may be patchy, leading to false-negative biopsies.
On imaging, findings in PACNS are highly variable and nonspecific. CT scan is abnormal in up to 67% of cases, and may reveal areas of low-density suggestive of ischemic events and, rarely, intraparenchymal or subarachnoid hemorrhage. MR imaging shows discrete or diffuse supratentorial and infratentorial lesions involving the deep and superficial white matter ( Fig. 1 ). In addition, areas of infarcts and hemorrhage may be seen (see Fig. 1 ; Fig. 2 ). On diffusion-weighted imaging (DWI), some acute/subacute lesions show restricted diffusion, suggesting an ischemic mechanism, whereas other acute/subacute lesions have higher apparent diffusion coefficient values, implicating a nonischemic mechanism. Post gadolinium, the lesions usually enhance in up to 90% of cases (see Figs. 1 and 2 ). In addition, enhancements can be seen in the Virchow-Robin spaces, leptomeninges, and spinal cord lesions (see Fig. 2 ). Patients with normal MR examination are unlikely to have abnormalities suggestive of angiitis on catheter angiography or brain biopsy. MR angiography is usually not informative, but may show vascular irregularities. Catheter angiography has a sensitivity and positive predictive value of less than 30%. It may show focal and/or multifocal segmental narrowing occlusion or irregularity of both small and medium-sized parenchymal and leptomeningeal blood vessels; collateral vessel formations, and prolonged circulation time (see Fig. 2 ; Fig. 3 ). Angiographically, PACNS cannot be differentiated from secondary causes of angiitis of CNS.
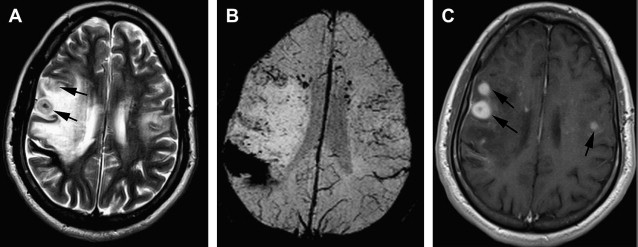
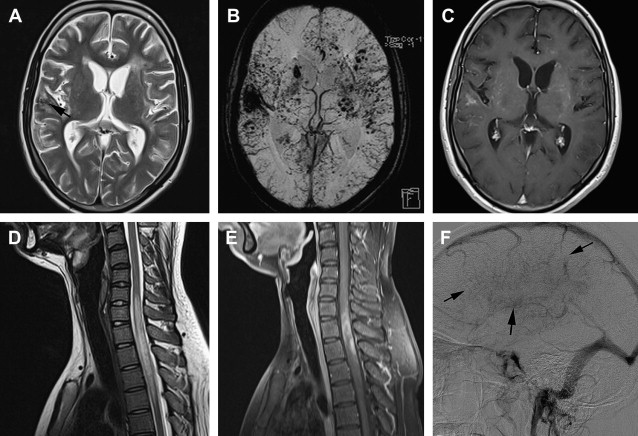
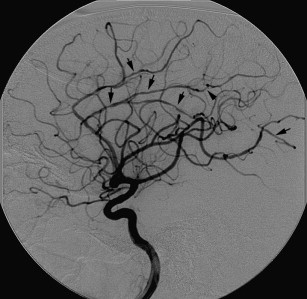
Histopathological confirmation by leptomeningeal and brain parenchymal biopsy remains the gold standard for the diagnosis of PACNS. This method is invasive, and findings may be negative in some patients with primary angiitis of the CNS because of sampling error. A negative biopsy does not rule out the condition. PACNS is managed with high-dose steroids and cytotoxic agents such as cyclophosphamide and azathioprine. Without treatment, the disease is usually fatal.
Giant cell arteritis
Giant cell arteritis (GCA) is a chronic, granulomatous vasculitis of large and medium-sized arteries. It involves the superficial temporal artery, but may also include other cranial arteries, for example, the occipital arteries. It usually occurs in patients older than 55 years. There are two common constellations of findings in GCA: temporal arteritis and polymyalgia rheumatica. Symptoms of temporal arteritis include unilateral headache, facial pain, jaw claudication, or loss of vision. Temporal arteritis is a common manifestation of GCA and may be confirmed by using temporal artery biopsy. Histologic analysis may reveal granulomatous inflammatory changes. In many patients, the clinical manifestation and laboratory values are suggestive of a diagnosis of temporal arteritis, and a biopsy is unnecessary. However, in cases of clinical uncertainty, biopsy may be required for a definitive diagnosis.
A characteristic gray-scale ultrasound finding of GCA involvement of the temporal artery is a diffusely thickened hypoechoic arterial wall or halo. Color and spectral Doppler ultrasonography may depict turbulent flow and stenosis of the affected vessel. Contrast-enhanced, high-resolution MR imaging may depict mural thickening and enhancement of vessel wall, thereby suggesting the diagnosis.
Secondary Vasculitis
Systemic vessel wall disease
Takayasu arteritis
Takayasu arteritis (TA) is an idiopathic chronic inflammatory disease affecting the aorta, its main branches, and the pulmonary arteries. TA leads to inflammation and fibrosis in the involved vessel walls resulting in luminal stenosis, occlusion, dilation, and aneurysm formation. This disease is also known as aortitis syndrome, nonspecific aortoarteritis, pulseless disease, atypical coarctation of aorta, and aortic syndrome, among others. Although reported worldwide, it is more frequently seen in Asia, the Mediterranean basin, South Africa, and Latin America. The annual incidence of TA in North America was previously estimated to be 2.6 per million. TA has overall female preponderance, although the female to male ratio seems to decline from eastern Asia toward the West. TA commonly occurs in the second and third decades of life.
The exact etiopathology remains unknown. The pathologic basis for this disease is a vasa vasoritis in the adventitia of large and medium-sized arteries. TA involves a continuous length of the vessel, producing mural and luminal changes in some areas, and only mural changes in the intervening segments. Even the angiographically normal areas have extensive wall changes on cross-sectional imaging.
The clinical manifestations of TA are usually divided into early and late phases. First there is an early or prepulseless phase characterized by nonspecific systemic features such as low-grade fever, night sweats, weight loss, arthralgia, and fatigue. The initial “inflammatory phase” is often followed by a secondary “pulseless phase” characterized by vascular insufficiency from intimal narrowing of the vessels manifesting as arm or leg claudication, renal artery stenosis causing hypertension, and neurologic symptoms secondary to renovascular hypertension or ischemia (postural dizziness, seizures, amaurosis, hemiparesis, aphasia, cranial nerve palsies, and vertigo).
Imaging features of acute phase
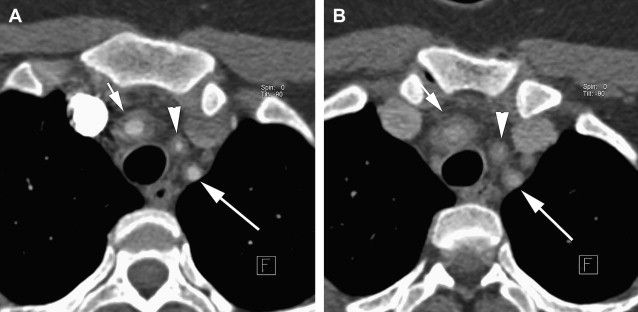
Significant findings of the acute phase of TA are wall thickening of the aorta and pulmonary artery. High-resolution B-mode ultrasonography can detect increased intimal-medial thickness (IMT), which is a reliable marker of active disease, especially in the absence of angiographically visible disease. Similarly, unenhanced CT scans may show a high-density wall of variable thickness in the aorta or its branches along with calcification, and enhanced CT scan may show enhancement of the thickened wall in patients with an active inflammation ( Fig. 4 ). MR imaging can show subtle wall thickening in early cases, and may show a bright signal due to edema in and around the inflamed vessel on T2-weighted images ( Fig. 5 ). During the acute phase, enhancement of the vessel wall and periadventitious soft tissues can also be observed (see Fig. 5 ). Delayed contrast-enhanced MR imaging may also be a useful technique to identify inflammation in the arterial wall. Overall these MR criteria are not highly sensitive, but are highly specific of disease activity. Sometimes occlusion of the aortic branches or pulmonary artery, or both, is seen in the acute phase. Rarely, pseudoaneurysm formation occurs in the acute phase. 18 F-Fluorodeoxyglucose positron emission tomography images coregistered with enhanced CT images have also demonstrated the distribution and inflammatory activity in the aorta, its branches, and the pulmonary artery in patients with active disease. The intensity of accumulation has been shown to decrease in response to therapy.
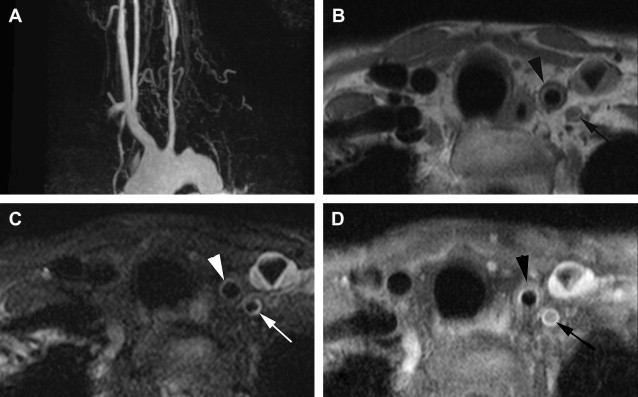
Imaging features of late phase
Significant findings of the late phase of TA include diffuse narrowing of the descending thoracic and abdominal aorta. Dilatation occurs most commonly in the ascending aorta. Stenosis, the most common finding, involves all arteries arising from the aorta, most commonly the common carotid and subclavian arteries (see Fig. 5 ; Fig. 6 ). Occlusion is the second most common finding. Abrupt occlusion, abrupt transition to collateral vessels, and flame-shaped termination are characteristic. Among branches of abdominal aorta, the renal artery is the most frequently involved. Pulmonary artery involvement is relatively high, with an estimated occurrence rate of 50% to 80%. TA is also associated with aortic dissection and pseudoaneurysm formation in the late phase. MR angiography depicts these findings well, and is also useful for evaluation of bypass graft. Cine MR imaging can depict aortic regurgitation caused by dilatation of the ascending aorta. Angiographic findings considered diagnostic of TA include a spectrum of changes ranging from minimal intimal irregularity to typical rat-tail narrowing, complete obstruction, or aneurysm formation in the involved vessels. Involvement of the aorta and/or at least 2 medium-sized branches is considered essential for diagnosis.
The diagnosis requires correlation of clinical, radiologic, and biochemical findings. The criteria for diagnosis include the presence of (1) symptoms caused by ischemia of the CNS, upper extremities, or kidneys; (2) fever, absent or decreased pulses, bruits, and fundoscopic findings; and (3) increased ESR and presence of C-reactive protein.
The goals of therapy in TA include the control of clinical activity by pharmacologic treatment using steroids and/or immunosuppressive therapy, restoration of blood flow to the stenosed vessel by surgical or endovascular techniques, pharmacologic control of blood pressure, and supportive management. Supra-aortic angioplasty is often not feasible in patients with TA because of involvement of the long segment of arteries. Surgical treatment can be offered in the form of a bypass graft (ascending aorta to carotid/subclavian). This repair is recommended in the “burnt-out” phase of the disease when further progression is unlikely.
Moyamoya disease
Moyamoya disease (MMD; moyamoya translates from Japanese as “puff of smoke”) represents a rare cerebral steno-occlusive disease characterized by progressive occlusion of the terminal segments of the internal carotid arteries (ICAs) and compensatory development of tortuous, dilated collateral networks. The result of these steno-occlusions is a severe impairment of the cerebrovascular reserve capacity. MMD is a progressive disease, as reflected by the MMD classification proposed by Suzuki and colleagues. The term moyamoya syndrome is used in cases in which an underlying cause (sickle cell anemia, Down syndrome, Marfan syndrome, tuberous sclerosis, Turner syndrome, von Recklinghausen disease, atherosclerotic disease, coarctation of the aorta, fibromuscular dysplasia, and tuberculosis) can be identified.
In MMD, vascular occlusion results from a combination of hyperplasia of smooth muscle cells and luminal thrombosis. The media is often attenuated with irregular elastic lamina. By definition, the pathognomic arteriographic findings are bilateral in MMD, although they can be asymmetric in degree and extent. Patients with unilateral findings have the moyamoya syndrome and eventually up to 40% develop contralateral disease during follow-up. Proximal posterior cerebral arteries may show stenosis or occlusion in up to 25% of patients with MMD.
The exact etiology of MMD is unknown. The disease may be hereditary and multifactorial and may occur in a previously healthy individual. Whereas most cases have been reported in East Asia thus far, the number of pediatric as well as adult cases of MMD reported within the Caucasian population is growing steadily, due to an increasing awareness of the disease. MMD has a bimodal age presentation, with the first peak occurring in the first decade of life and second peak in the fourth decade. Among pediatric patients, ischemic symptoms are the most common presentation, whereas adults may present with either ischemic or a hemorrhagic stroke, or both. In the United States, the rate of hemorrhage among adults is approximately 7 times as high as the rate among children (20% vs 2.8%). Compared with adults in the United States, adults in the Asian population have a much higher (42%) rate of hemorrhage in MMD.
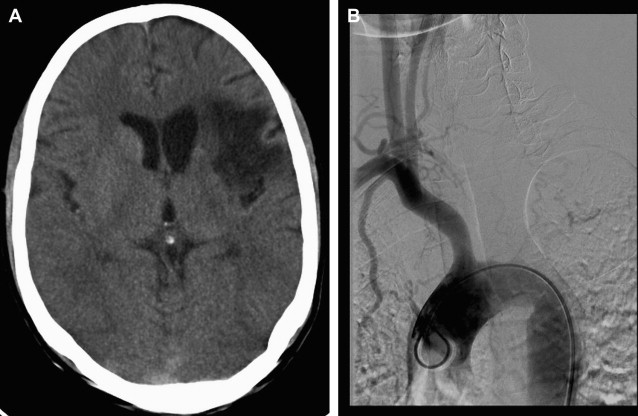
Depending on presentation, imaging may show evidence of ischemia (acute, subacute, or chronic) or hemorrhage (parenchymal, intraventricular or, rarely, subarachnoid). MR imaging can reveal stenosis or occlusion of distal ICA, decrease in size of flow voids of middle cerebral arteries (MCAs) and anterior cerebral arteries, presence of moyamoya vessels with signal voids within the basal ganglion and thalami, as well as ischemia, infarction, atrophy, and ventriculomegaly ( Fig. 7 ). T2∗-weighted MR images may detect asymptomatic microbleeds in about 15% to 44% of adult patients. On fluid-attenuated inversion recovery (FLAIR) images, diffuse leptomeningeal collaterals may appear as sulcal hyperintensity, referred to as the ivy sign (see Fig. 7 ). This ivy sign is seen in hemisphere with decreased cerebrovascular reserve. Diffusion-weighted and perfusion imaging are useful for evaluating cerebral ischemia in MMD. Regional cerebral perfusion in patients with MMD is decreased and delayed with posterior cerebral artery (PCA) stenosis, with a greater decrease and delay with PCA occlusion. If the PCA is occluded, the number of leptomeningeal collateral vessels to the anterior circulation presumably decreases; thereby causing severe cerebral ischemia that results in an infarction. Contrast-enhanced T1-weighted images show marked leptomeningeal enhancement along the cortical sulci, as well as enhancement of moyamoya vessels in cases with advanced MMD.
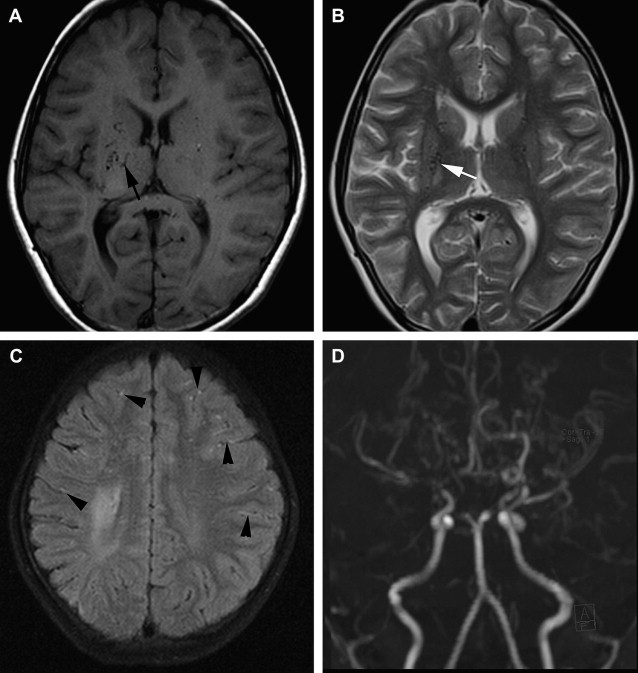
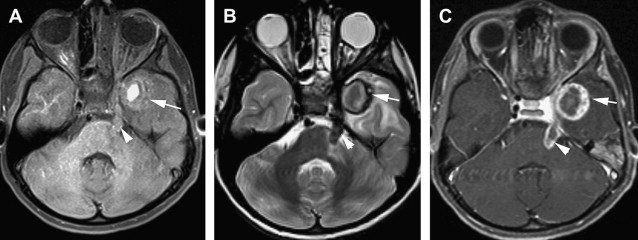
MR angiography is also useful for the diagnosis of MMD in a noninvasive way. MR angiography shows terminal ICA stenosis or occlusion, and the presence of moyamoya collaterals (see Fig. 7 ). Conventional arteriography, the ultimate gold standard in the evaluation of patients suspected to have MMD, is reserved for presurgical evaluation before vascular anastomosis and is also useful in assessing the development of collateral pathways following surgical revascularization ( Fig. 8 ). Incidental major artery aneurysms can be found in 3.6% of adult patients with nonhemorrhagic MMD, an observed frequency that increases with age. About half of these aneurysms are located in the posterior circulation, particularly in older patients with bilateral MMD ( Fig. 9 ).
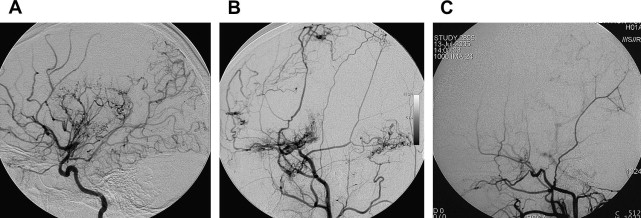
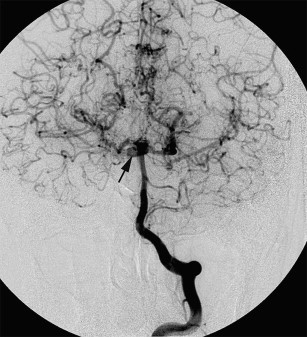
Patients with MMD seem to progress with time, even if they are asymptomatic. Surgical treatment has been advocated as the treatment of choice to improve cerebral hemodynamics and to reduce the risk of subsequent strokes. Three types of surgical revascularization procedures can be done: direct (superficial temporal artery-MCA anastomosis), indirect (encephaloduroarteriosynangiosis, encephalomyosynangiosis, and so forth), or combined.
Polyarteritis nodosa
Polyarteritis nodosa (PAN) is a focal panmural necrotizing vasculitis in small and medium-sized arteries (and sometimes small veins) that can involve any organ and in varying degrees. The kidneys may be involved in 70% to 80% of cases; the gastrointestinal tract, peripheral nerves, and skin in 50%; skeletal muscles and mesentery in 30%; and the CNS in 10%. The characteristic pathologic findings are fibrinoid necrotizing inflammatory foci in the walls of small and medium-sized arteries, with multiple small aneurysms ( Fig. 10 ). In the reparative and chronic stages, fibroblast proliferation causes wall thickening and may produce areas of stenosis or occlusion, which may lead to ischemia or infarction. This pathophysiology produces the two angiographic findings of polyarteritis nodosa, aneurysms on the one hand and stenoses or occlusions on the other. If left untreated, polyarteritis nodosa is usually fatal. However, treatment with corticosteroids and cyclophosphamide improves the chance for survival.
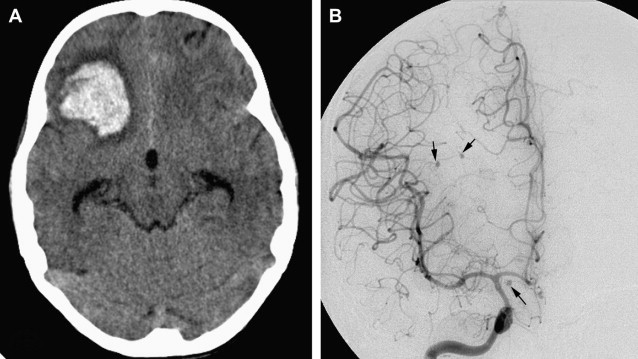
Autoimmune vasculitis
Systemic lupus erythematosus, Behçet disease, Wegener granulomatosis, and Sjögren syndrome comprise the majority of autoimmune conditions associated with CNS vasculitis. In all of these disorders, the precise pathogenesis remains obscure; in all, however, the immune system seems to play a central role, and immunosuppressive agents are the cornerstones of treatment.
On MR imaging of brain the typical appearance of CNS vasculitis includes multiple subcortical infarctions, although substantial variation in the number, size, and location of the lesions may occur in this condition. MR imaging may not reveal all abnormal brain regions in such patients. MR studies reveal abnormalities in the vast majority of patients with CNS autoimmune vasculitis, and in this sense the sensitivity of MR imaging is quite high. However, findings on MR imaging are not specific for vasculitis, and similar MR findings may occur in patients suffering from multiple sclerosis, low-grade gliomas, mitochondrial disorders, substance abuse, and other conditions.
Table 1 contains a brief summary of clinical findings and imaging features in various autoimmune vasculitides.
| Disease Name | Systemic Diagnosis | Neurologic Symptoms | Laboratory Findings | Imaging | |
|---|---|---|---|---|---|
| 1 | Rheumatic Disease | Severe constitutional symptoms Prominent extra-articular manifestations of vasculitis affecting the skin, eyes, heart, and/or lungs | Nonspecific multitude of neurologic signs and symptoms | Rheumatic factor Elevated ESR | Scattered WM lesions |
| 2 | Systemic Lupus Erythematosus (SLE) | Often presents with neuropsychiatric symptoms | Antinuclear antibodies Antibodies against dsDNA | Cortical infarctions mimicking MELAS Scattered WM lesions mimicking multiple sclerosis Severe basal ganglion changes mimicking deep venous thrombosis Extensive calcification of the basal ganglion, dendrate nucleus, centrum semiovale, and corticosubcortical region Normal MR imaging | |
| 3 | Sneddon syndrome | Generalized livedo recemosa | Large and small arterial infarcts with severe neurologic consequences and often cognitive deterioration | Elevated ESR Anticardiolipin antibodies | Multiple smaller and large infarcts with prominent cortical involvement Multiple hemosiderin deposits with extensive superficial hemosiderosis |
| 4 | Antiphospholipid antibody syndrome (1° or 2° to SLE, postinfections drug induced) | Hypercoagulative state, which leads to thrombotic complications, stroke, myocardial infarctions, dementia, and fetal loss. Cardiac value vegetations, mitral regurgitation | Infarcts Early-onset dementia | Multiple WM lesions, lesion usually extends into the cortical layers | |
| 5 | Sjögren syndrome | Involvement of salivary and lachrymal glands; constitutional symptoms: fatigue, malaise, low-grade fever; Reynaud syndrome; lymphadenopathy | Trigeminal neuropathy Recurrent aseptic meningoencephalitis Transverse myelitis Chronic progressive myelitis Neuropsychiatric symptoms | Nonspecific extensive gray and white matter lesions Microhemorrhages | |
| 6 | Scleroderma | CREST (Calcinosis, Reynaud syndrome, Esophageal problems, Sclerodactylia, and Telangiectasia syndrome) | Pain and parethesias from nerve entrapment; territorial strokes, epileptics seizures, recurrent loss of consciousness, multiple spontaneous intracerebral hemorrhages | Anticentromere antibiotics | Nonspecific Middle-sized artery infarcts Macrohemorrhages and microhemorrhages Extensive calcifications |
| 7 | Neurosarcoidosis (5% of patients with systemic sarcoidosis) | Systemic granulamatous disease, affecting lungs with lung fibrosis, hilar lymph nodes skin, liver, spleen | Facial nerve paralysis (most common manifestation) | Not very helpful Positive angiotensin-converting enzyme test in CSF pleocytosis | Suprasellar leptomeningeal disease WM lesions |
| 8 | Wegener granulomatosis | Recurrent epistaxis or sinusitis, pulmonary infiltrates and/or nodules, glomerulonephritis, ocular involvement Dyspnea, hoarseness, cough, wheezing, and other symptoms of tracheal or parenchymal lung diseases | Cranial neuropathy Seizure Peripheral neuropathy, stroke, or TIAs Headache | Cytoplasmic antineutrophil antibodies in 90% | Sinusitis Destruction of petrous part of temporal lobe Erosion of lamina papyracea Enhancement of cranial nerves Pachymeningeal thickening and enhancement Infarct |
| 9 | Churg-Strauss syndrome, granulamatous inflammation, and necrotizing vasculitis involving small to medium-sized vessels | Allergic rhinitis, asthma, eosinophilia, pulmonary infiltrates, coronary arteritis, intestinal ischemia | Commonly involves the peripheral nervous system and presents as a mononeuritis multiplex CNS involvement is seen in 10% of patients | Perinuclear antineutrophil antibodies (pANCA) | Macroinfarctions or microinfarctions Macrohemorrhages or microhemorrhages |
| 10 | Microscopic polyarteritis (polyangiopathy), necrotizing vasculitis of small vessels, arterioles, capillaries, or veins without granulomas | Pulmonary hemorrhage, glomerulonephritis | Peripheral and cranial nerve involvement Stroke | pANCA (+vein 50%–80%) | Macroinfarcts or microinfarcts |
| 11 | Behçet disease (multisystem recurrent vasculitis of supposedly autoimmune origin); blood vessels of all sizes are involved, arteries as well as veins | Clinical triad: recurrent oral ulcers, genital ulcers, and anterior or posterior uveitis | CNS involvement in 5%–10% of cases Headache Meningoencephalitis Cranial nerve palsies Cerebellar ataxia Spastic paraparesis or tetraparesis Raised intracranial pressure Dementia | CSF pleocytosis, elevated protein, and elevated pressure | Asymmetric mesodiencephalic junction lesion with edema extending along certain long tracts in the brainstem, sparing red nucleus and involving diencephalon, pontobulbar region lesions; brainstem lesions are more frequent, larger, and more extensive than in multiple sclerosis Hemispheric WM lesions: focal or confluent, more likely to be subcortical than periventricular Spinal cord lesion: posterolateral, mainly cervical cord Cranial nerve enhancement, enhancement of intracranial and spinal cord lesions Cerebral venous thrombosis |
Infectious vasculitis
Infection is a well-recognized cause of secondary vasculitis; many different agents have been implicated. It is important to have a high index of suspicion for infection as the cause of vasculitis, because treatment can be directed at the underlying organism. General clues to infectious vasculitis are associated fever, abnormality of the peripheral leukocyte count, and recent or current extraneural infection.
Bacteria
Acute septic meningitis, attributable to several bacterial agents, may cause vasculitis and cerebral infarcts in 5% to 15% of adults with bacterial meningitis and in up to 30% of neonates.
Mycobacterium tuberculosis is the commonest cause of chronic meningitis, and typically follows rupture of an old tubercle with the release of mycobacteria in the subarachnoid space. The thick gelatinous inflammatory exudate that typically contains organisms, mononuclear cells, tubercles, and caseation necrosis settles at the base of the brain along cisterns, particularly in the Sylvian fissure, where arteritis can form along traversing blood vessels. Resultant ischemic infarction occurs in up to 41% of patients. The infection may include vasculitis of the smaller and middle-sized cerebral arteries, often the lenticulostriate arteries or the posterior cerebral branches and the thalamoperforating arteries, leading to small infarctions in the basal ganglion and deep white matter ( Fig. 11 ).
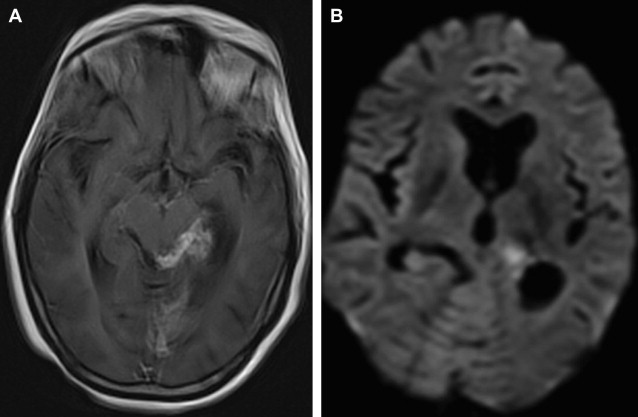
Fungi
Four fungal agents, Aspergillus , Candida , Coccidioides , and Mucormycetes , are important CNS pathogens, particularly in the setting of leukopenia, sepsis, drug-induced or human immunodeficiency virus (HIV)-related immunosuppression, and severe debilitation. All have the capacity to invade arteries of the CNS in the course of disseminated infection and meningitis. Vasculitis may be acute, subacute, or a late complication of CNS infection. Cerebral infarction results from direct vascular injury leading to aneurysm formation, vascular thrombosis, endarteritis, and cerebral hemorrhage, or results from microabscesses, or extension along contiguous sites of infection. Mucormycosis may be particularly aggressive in poorly controlled diabetes. There may be associated spread from sites of nasopharyngitis, oropharyngitis, and sinusitis to the cavernous sinus and ICA, leading to focal thrombosis and cerebral infarction, detectable by imaging studies ( Fig. 12 ).