derivatives of cytotoxic T cells and that nodular lymphocyte-predominant HL (NLPHL) is a distinctive and uncommon lymphoproliferative disorder. The lymphocytic and histiocytic (L&H) cells characteristic of NLPHL have folded and lobate nuclei among small lymphocytes and histiocytes. The L&H cells are also B cell-derived but harbor V gene alterations, which differ from those in HRS cells; whether the L&H cells are monoclonal is unclear (34,35). The clinical characteristics of NLPHL differ from those of the subtypes of CHL by virtue of its indolence, excellent prognosis, epidemiology, and response to chemotherapy. The Revised European-American Classification of Lymphoid Neoplasia (REAL) has devised a schema that reflects the distinction of NLPHL from CHL (36).
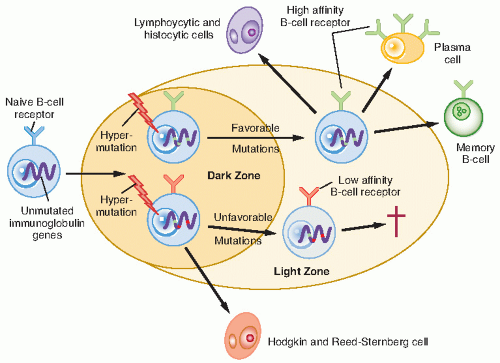 Figure 7.1 The derivation of Hodgkin and Reed-Sternberg cells in classic Hodgkin’s lymphoma and lymphocytic and histiocytic cells in nodular lymphocyte-predominant Hodgkin’s lymphoma. (From Thomas RK, Re D, Wolf J, et al. Part 1: Hodgkin’s lymphoma- molecular biology of Hodgkin and Reed-Sternberg cells. Lancet Oncol 2004;5:11-18.) |
associated with inferior OS in patients greater than the age of 50 years when adjusting for stage, sex, and B symptoms, although it was not associated with inferior disease-specific survival (48). Analysis of the database of the International Hodgkin Study Group revealed no significant differences associated with EBV positivity in failure-free survival or OS after adjustment for stage and age (45,49). Understanding the pathogenesis and prognostic implications of EBV-associated HL may help to improve immune-directed therapeutic strategies.
Table 7.1 Ann Arbor Staging System with Cotswold Modifications for Hodgkin Lymphoma | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
small number of cells present in the specimen, and the difficulty of classifying HL into one of the four categories of the Rye classification. The Rye classification subcategorizes HL into nodular sclerosing (NSHL), MCHL, LPHL, and lymphocyte-depleted (LDHL) types. With modern treatment the prognostic significance of these subtypes has diminished, although the presenting characteristics and natural history remain evident, particularly for the nodular subtype of LPHL. The importance of distinguishing NLPHL from the other types has led to the REAL classification (Table 7.2) (39). The clinicopathologic characteristics are the same as those in adults and are briefly described here.
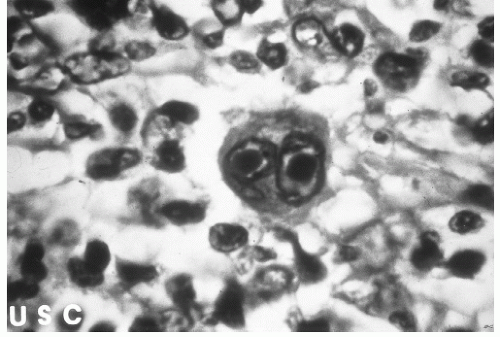 Figure 7.2 Reed-Sternberg cell. |
NLPHL (58): The distinctive cell is the L&H “popcorn” cell, which is CD20 (B-lymphocyte marker) positive and CD 15 negative. Classic HRS cells (which are usually CD15 and CD30) are rare, as is the detection of EBV. Progressive transformation of the germinal centers of lymph nodes is often seen, and, in fact, can occur in the absence of NLPHL. Therefore, it is important to distinguish these entities. NLPHL has a long natural history, in its time to diagnosis and to relapse, reminiscent of that of indolent non-Hodgkin lymphomas (NHL). It is more common in young children (33% of all patients are younger than 15 years), has a high male:female ratio (4:1), and commonly involves a single lymph node region with sparing of the mediastinum (59).
Lymphocyte-rich (classic) HL: HRS cells (CD15) are identifiable against a background predominantly of lymphocytes. Clinical behavior is similar to that of MCHL.
Mixed-cellularity (classic) HL: HRS cells (CD15) are common against a background of abundant normal reactive cells (lymphocytes, plasma cells, eosinophils, histiocytes). This subtype can be confused with peripheral T-cell NHL. MCHL is less common in children, often accompanied by “B” symptoms, and more often involves infradiaphragmatic nodes.
Nodular sclerosis (classic) HL: This subtype is distinctive because of the presence of collagenous bands that divide the lymph node into nodules, which often contain an HRS cell variant called the lacunar cell. NSHL often occurs in children, involves supradiaphragmatic nodes, and spreads in an orderly manner along contiguous nodal chains.
Lymphocyte-depleted (classic) HL: This subtype is rare and commonly confused with NHL, particularly of the anaplastic large cell type. HRS and pleomorphic variants are common relative to the number of background lymphocytes. LDHL often is advanced at diagnosis and has a poor prognosis.
Table 7.2 Comparison of Revised European-American Classification of Lymphoid Neoplasia (REAL) and Rye Classification of the Hodgkin Disease | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
subclassifications of the Ann Arbor staging system have been proposed, particularly for patients with large mediastinal adenopathy (LMA) or stage IIIA disease. LMA, most commonly defined as a mass exceeding one third the transverse diameter of the chest (intrathoracic width measured at the dome of the diaphragm) on a standard upright posteroanterior chest radiograph, places a patient at a greater risk for disease recurrence after radiation alone. Patients with pathologic stage (PS) III disease limited to the spleen or splenic, celiac, or portal nodes are denoted anatomic substage III1 and considered to have a more favorable prognosis than patients with involvement of paraaortic, iliac, or mesenteric nodes, denoted as III2 (60). However, this system has not proven useful in some centers.
Table 7.3 Demographic and Clinical Characteristics at Presentation of Pediatric Hodgkin Disease | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
diseases (64). Patients with “B” symptoms or stage III-IV disease should undergo bone marrow biopsy (65). The low yield of a bone marrow evaluation in asymptomatic patients with localized disease (stage I and II) does not support routine use of the procedure.
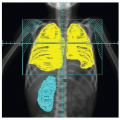
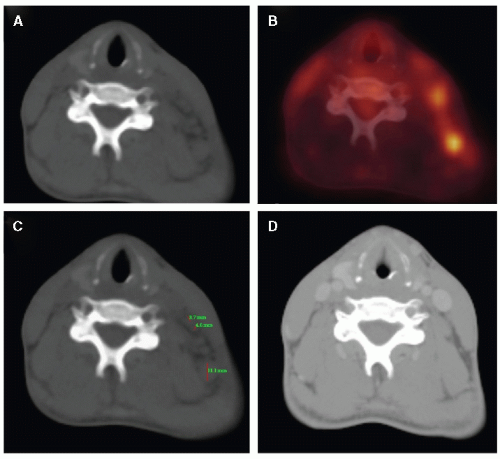 Figure 7.3 A: A few poorly identifiable lymph nodes in the left cervical area on an axial CT slice without IV contrast before chemotherapy. B: FDG-PET image of the same area. C: Measurements of the largest lymph nodes (3.7, 4.6 and 11.1 mm). D: The left cervical area after chemotherapy. |
rebound commonly observed after completion of therapy for HL).
and 84% and 73% for adults (20). Although children younger than 4 years with HL are uncommon, even these children appear to have an excellent prognosis (21).
For patients with PHL, is chemotherapy alone adequate without administering excessive amounts or using overly aggressive combinations in terms of potential morbidities? Embedded in this question are the relative toxicities of curative chemotherapy versus less chemotherapy plus RT.
Can the rapidity and the completeness of the response to chemotherapy define the use of RT, and will functional imaging enhance the precision of this approach?
When RT is administered, then what is the appropriate target volume? Can RT be restricted to initially involved lymph nodes rather than chains (or regions) of nodes? In what settings should RT be directed to areas of initial bulk disease or residual postchemotherapy disease? Should involved organs, such as the liver, lung, and heart, ever be irradiated?
If we use RT, what is the appropriate dose? Should this be dependent on the initial or postchemotherapy extent of disease, bulk of disease, organ at risk, and age of the patient?
Table 7.4 Treatment Results for Early, Favorable Pediatric Hodgkin Disease | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 7.5 Treatment Results for Advanced, Unfavorable Pediatric Hodgkin Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


