Abstract
Holoprosencephaly (HPE) is a heterogeneous central nervous system (CNS) anomaly that results from a primary defect in induction and patterning of the rostral neural tube (basal forebrain), leading to varying degrees of incomplete separation of the cerebral hemispheres and facial anomalies. HPE is graded as alobar, semilobar, and lobar, and a mild version called middle interhemispheric variant of HPE . The variable facial anomalies are: (1) cyclopia, with or without proboscis; (2) ethmocephaly with ocular hypotelorism and proboscis located between the eyes; (3) cebocephaly with ocular hypotelorism and single-nostril nose; and (4) median cleft lip/palate (premaxillary agenesis) and ocular hypotelorism.
The prevalence of severe HPE in embryonic population is approximately 50 : 10,000, whereas in nonselected fetal population it is approximately 1.26 : 10,000. HPE study populations differ depending on the age of the specimens, the time of the detection of HPE, the number of cases, identified subgroups (e.g., chromosomal, nonchromosomal, not karyotyped), and the type of population (selected or nonselected).
There are various etiological factors involved in HPE (environmental and chromosomal factors, genetic syndromes). Depending on the etiology , a wide range of associated anomalies have been reported.
US remains the primary imaging modality for examination of the fetal brain and thus HPE. MRI can be an important supplement in suspected fetal HPE, especially in the middle interhemispheric variant of HPE.
There is a high prenatal and perinatal mortality rate of severe cases of isolated HPE. No treatment options are available. Surviving children with HPE have developmental disabilities correlating with the severity of brain malformation.
Keywords
alobar, holoprocencephaly, lobar, middle interhemispheric variant, semilobar
Introduction
Holoprosencephaly (HPE) has been known since antiquity through the figure of the cyclopean shepherd Polyphemos in Homer’s Odyssey (circa 800 bc ). Until the 17th century, a cyclopic newborn, whether it was a human or an animal, was associated with mystic and fabulous narrations. In the 18th century, HPE and other anomalies were recognized as congenital conditions, and cases of HPE were collected and described scientifically. Recently, a 1500-year-old skull of a 5-year-old child (based on ossification and tooth eruption) who had a premaxillary agenesis and ocular hypotelorism was found in Peru.
Recognition of HPE prenatally by two-dimensional (2D) ultrasound (US) was first described by Kurtz et al. in 1980 and has since been described in many case reports and studies. Alobar HPE is the most severe form of HPE and can be diagnosed with 2D and three-dimensional (3D) US in the embryonic period at 9 weeks’ gestational age. Although magnetic resonance imaging (MRI) was introduced into fetal imaging in 1984, and fetal HPE was described by MRI in 1991, this imaging modality still has limited application in diagnosis of fetal HPE because MRI investigations require resources. Neuroimaging studies of postnatal HPE are mainly based on MRI and have shown an extensive variety of expression. The full range of the variety of expression of HPE and associated anomalies has not yet been described in prenatal studies.
Disease
Definition
HPE is a heterogeneous central nervous system (CNS) anomaly that results from a primary defect in induction and patterning of the rostral neural tube (basal forebrain), leading to varying degrees of incomplete separation of the cerebral hemispheres. The categorization of HPE introduced by DeMyer and Zeman in 1963 and modified by DeMyer in 1977 is still generally accepted. HPE is graded according to the severity of the brain anomaly as alobar, semilobar, and lobar . A mild version of HPE called middle interhemispheric variant of HPE has also been identified.
Prevalence and Epidemiology
Embryo
In 2004, a large embryonic study of HPE from Japan was published. Embryos were diagnosed with HPE if they had characteristic craniofacial features, such as closely apposed or fused eyes, proboscis, and abnormally narrow heads. They found 201 HPE cases among 44,000 human embryos ranging from Carnegie stages 13 to 23, which corresponds approximately to 6 to 10 completed weeks based on last menstrual period. The prevalence of severe HPE in this embryonic population was 50 : 10,000. The actual prevalence is probably higher because mild forms of HPE could have escaped detection, particularly in the early stages of craniofacial development. This Japanese study did not classify the types of HPE according to brain anomalies, and karyotype was unknown.
Fetus
Many US reports describe the diagnosis of fetal HPE. A few fetal studies allow some epidemiologic conclusions. Kagan reported 57,119 pregnancies at 11 to 13 weeks’ gestation, in which 44 cases of alobar HPE were detected, a prevalence of alobar HPE of 7.7 : 10,000. Of the 44 cases, 29 (66%) were aneuploid. Two likely reasons account for the high prevalence in this study: (1) early pregnancy losses in HPE fetuses, especially among fetuses associated with presumptive lethal chromosome defects, would not appear in series ascertained at birth, and (2) this may have been a selected population.
Blaas et al. reported 17 fetuses from a referral population and 13 fetuses from a nonselected population of 119,411 deliveries (greater than or equal to 16 weeks’ gestation). The mean age of detection of the cases was 21 ± 3 weeks’ gestation. After reviewing records at all hospitals with departments of obstetrics, pediatrics, and pathology within the catchment area, two further HPE cases (one alobar and one semilobar) were identified that had not been diagnosed before delivery and resulted in death shortly after birth. Cases with lobar HPE were not identified. The prevalence of HPE in the nonselected fetal population was 1.26 : 10,000. Chromosome aberrations were present in 37% (11 of 30) of the whole study group. The brain anomalies consisted of 18 lobar HPE (60%), 5 semilobar HPE (17%), 4 lobar HPE (13%), and 3 anencephalic HPE (10%; see later).
A study from Taiwan was based on HPE cases that were detected by US among cases referred for fetal karyotyping (Chen C, personal communication). A high prevalence of 6 : 10,000 could be the result of selection bias of pregnancies at risk for abnormal chromosomes. Only alobar HPE types were diagnosed, and 34 (58%) had abnormal chromosomes.
A perinatal study from California (121 HPE diagnoses in 1,035,386 live births and fetal death deliveries, registered from greater than or equal to 20 weeks’ gestational age to 1 year after delivery) reported a prevalence of 1.2 : 10,000. The investigators found 56 alobar (46%), 24 semilobar (20%), and 11 lobar (9%) HPE cases, and 30 specimens (25%) had undetermined HPE type.
International Studies
Large international studies including termination of pregnancies, stillbirths, and live births have found relatively high prevalences of HPE. The prevalence of HPE cases ascertained was 2.16 : 10,000 in a hospital-based population of 179 cases from South America (during 2000 to 2003). In the International Clearinghouse Birth Defects Surveillance Registry, the prevalence was 1.31 : 10,000, based on 963 HPE cases from 2000 to 2004. A similar prevalence of 1.33 : 10,000 was observed in the EUROCAT registry in 2010, which included 827 cases of arrhinencephalia or HPE in approximately 6,218,000 deliveries occurring from 2000 to 2008.
HPE study populations differ depending on the age of the specimens, the time of the detection of HPE, the number of cases, identified subgroups (e.g., chromosomal, nonchromosomal, not karyotyped), and the type of population (selected or nonselected). Substantial variations also exist in the identified cerebral defects and accompanying facial anomalies that generate differences in the ascertainment and categorizing of HPE cases. The discrepancy between embryonic and early fetal populations versus perinatal populations indicates a high loss rate of fetuses with HPE early in the first trimester. In addition, it is probable that some HPE cases are not recognized perinatally and not registered. Newborns with HPE and normal faces and infants with mild cerebral HPE types are especially prone to being missed. The inclusion of diagnoses until age 1 year in the postnatal study in California probably explains the relatively high prevalence and large share of mild HPE types. HPE features in conditions such as anencephaly and agnathia can also be overlooked or ignored in some studies.
Etiology and Pathophysiology
Etiologic heterogeneity contributes to the noticeable differences among HPE studies. Environmental, genetic, multifactorial, and unknown causes seem to be involved in the origin of this condition. HPE is an example of a multifactorial trait that may require synergy between mutations in key genes, interaction of these mutations with endogenous variants, and additional environmental effects.
Embryology
A common denominator for the large variety of the HPE spectrum is a defective induction of the prosencephalic neuroepithelium, including neural crest population by the prechordal plate during embryonic development, which may lead to HPE with its variable facial and cerebral appearances. The mesendodermal prechordal plate is usually detectable in human embryos at Carnegie stages 7 and 8 rostral to the notochordal process in contact with the floor of the neural groove. At stages 9 and 10, the plate is related to neuromere D1, which consists of thick neural ectoderm and comprises the optic primordium. In humans, the critical time period for teratogenic induction of HPE is in “the [embryologic] 3rd to early 4th week” of human development, which corresponds approximately to Carnegie stages 9 and 10, or 4 weeks, 6 days to 5 weeks, 2 to 3 days, based on the last menstrual period.
Environmental Influences
Maternal factors such as diabetes have been associated with HPE. According to a study from 1983, the approximate risk to infants of diabetic mothers is 1% to 2%, which is a 200-fold greater risk than the general population. The pathogenetic factors and their effects could be summarized as the result of any interference with the glycolytic pathway leading to a decreased rate of glycolysis and conversion of glucose to pyruvate.
Animal studies have revealed that numerous teratogens may be involved in the development of HPE. In a review of teratogenesis of HPE, Cohen and Shiota summarized that various factors may be involved in the development of HPE, such as maternal alcohol consumption during early pregnancy, the influence of isotretinoin and etretinate on neural crest cells, mutated genes, and teratogens involving the Sonic Hedgehog (SHH) signaling pathway including perturbations in cholesterol homeostasis, which is an important part of this pathway. Two known causes of HPE in humans, fetal alcohol exposure and maternal diabetes, are both associated with elevated levels of reactive oxygen species. The possible involvement of oxidative stress in the pathogenesis of HPE was shown by treating pregnant mice with antioxidants (vitamins C and E) to reduce oxidative stress. They partly prevented ethanol-induced apoptosis in the prechordal mesendoderm and HPE malformations. This study may have important implications to the pathogenetic mechanisms and gene-environmental interaction in the genesis of HPE.
Abnormal Karyotype
The most frequently identified etiology of HPE is an abnormality of chromosome numbers. These abnormalities include trisomy 13, trisomy 18, and triploidy, although several other aberrations have been reported. Such chromosome aberrations are almost universally fatal in gestation or in infancy. Trisomy 13 accounts for 75% of HPE cases caused by chromosomal anomalies (including cryptic rearrangements). Triploidy accounts for 20% of HPE cases. Trisomy 18 is less commonly seen in conjunction with HPE and accounts for 1% to 2% of the cases. HPE has also been reported in patients with trisomies other than trisomies 13 and 18. Overall, the recurrence risk of HPE secondary to cytogenetic anomalies is approximately 1%.
Previous research on the genetic etiology of HPE showed at least 12 HPE candidate loci on 11 chromosomes, designated HPE 1 through 12. Advances in cytogenetic techniques have enabled higher resolution and improved descriptions of chromosome aberrations in cases with HPE and have identified at least 14 genes where heterozygous mutations cause HPE, and even suggest that in some families several genetic events are necessary to cause HPE.
Syndromes and Associations
HPE may be associated with numerous syndromes such as Meckel syndrome, Martin syndrome, Fitch syndrome, Pallister-Hall syndrome, Steinfeld syndrome, hypertelorism and ectrodactyly syndrome, velocardiofacial syndrome, pseudo–trisomy 13 syndrome, Lambotte syndrome, Genoa syndrome, Goldenhar syndrome, acrocallosal syndrome, and Smith-Lemli-Opitz syndrome. Smith-Lemli-Opitz syndrome is due to a deficiency in the final step in the cholesterol biosynthetic pathway, 3β-hydroxy-steroid-Δ -reductase, and cholesterol is an essential part of the SHH signaling network.
Associated Anomalies
Depending on the etiology, either chromosome aberrations or nonchromosomal syndromes, a wide range of associated anomalies have been reported.
Manifestations of Disease
Clinical Presentation
Brain.
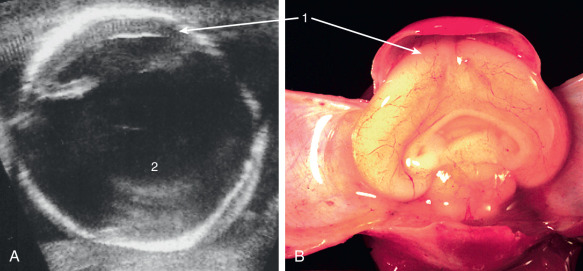
In alobar HPE, only a single ventricle (holosphere) of variable size is present, lacking interhemispheric division (see – ). Posteriorly, a membrane usually closes the holosphere. This membrane may bulge dorsally and form a fluid-filled “dorsal sac” ( Fig. 39.1 ). The thalami and corpora striata are undivided across the midline; the olfactory tracts and bulbs and the corpus callosum are always absent ( Fig. 39.2 ).
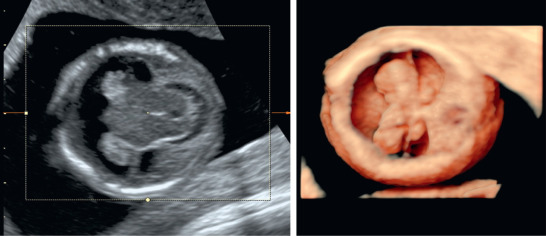
In semilobar HPE, there are rudimentary cerebral lobes, and an interhemispheric fissure may be present posteriorly ( Fig. 39.3 ). The olfactory tracts and bulbs are absent or hypoplastic, and the corpus callosum is rudimentary.

In lobar HPE, some midline continuity may be present despite a distinct interhemispheric fissure. The olfactory tracts and bulbs may be absent, hypoplastic, or normal. Midline cleavage of the thalami and the corpora striata may be incomplete.
In the middle interhemispheric variant of HPE, the posterior frontal and parietal lobes fail to separate. Hahn and Barnes presented a detailed overview of the neuroimaging features of HPE based on postnatal high-resolution brain MRI studies ( Table 39.1 ).
The HPE spectrum extends further into microforms with interhemispheric separation and only subtle midline brain defects (e.g., agenesis of corpus callosum). Microforms may have normal brains by conventional neuroimaging, but display subtle craniofacial anomalies consistent with a midline defect, including hypotelorism (closely spaced eyes), a flat or sharp nasal bridge, choanal stenosis, or a single maxillary central incisor (SMCI). Recently Solomon et al. presented five cases of microform HPE, four of whom had identified mutations in HPE-associated genes, who all had evidence for above-normal intelligence. This report expands the phenotypic spectrum of holoprosencephaly and is important in the counseling of patient and affected families.
Face.
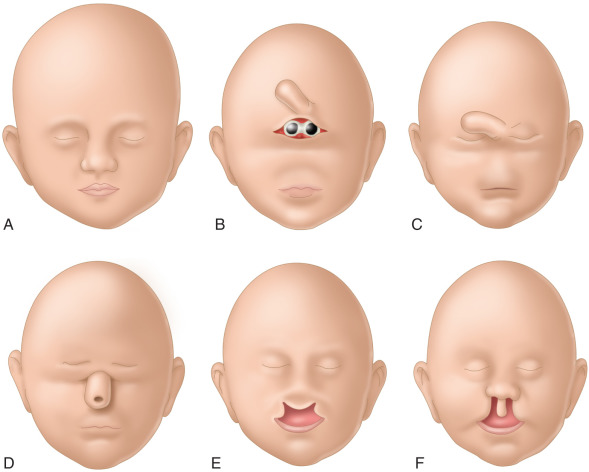
The primary defect in induction and patterning of the rostral neural tube (basal forebrain, neural crest cells) also leads to variable facial anomalies ( Fig. 39.4 ), which usually are categorized in four main types: (1) cyclopia with a single eye or various degrees of doubling of the eye anlage, with or without proboscis ( Fig. 39.5 ); (2) ethmocephaly with ocular hypotelorism and proboscis located between the eyes; (3) cebocephaly with ocular hypotelorism and single-nostril nose ( Fig. 39.6 ); and (4) median cleft lip/palate (premaxillary agenesis) and ocular hypotelorism ( Fig. 39.7 ).