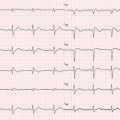
Fig. 9.1
Histologic specimen (Azan-Mallory, ×25) of a patient with hypertrophic cardiomyopathy (HCM) showing myocardial fiber disarray and interstitial fibrosis
9.1.1 Epidemiology
HCM is a disease widespread worldwide, with similar prevalence reported in different countries: 1:500 individuals [2]. It can be diagnosed throughout life, from childhood to old age, but it is most commonly seen for the first time among young adults. The disease shows rapid, sometimes exponential, phenotypic onset and expression throughout adolescence, in parallel with growth, indicating close noninvasive clinical and echocardiographic screening during this period. This applies above all to individuals with a family history or known to be carriers of a sarcomeric-causing disease mutation (genotype-positive/phenotype-negative individuals). Probably, many cases of HCM remain undiagnosed because of its asymptomatic course and normal life expectancy.
9.1.2 Genetics, Pathogenesis, and Pathophysiology
HCM is considered a genetic disease and is generally transmitted with an autosomal dominant pattern, shows a variable penetrance, and has great variability in phenotypic expression. Familial HCM accounts for 50 % of cases, with the remaining sporadic cases assumed to be de novo mutations [3]. The disease is caused by a singular mutated allele in one of more than 11 genes encoding for sarcomeric contractile myofilament proteins or components of the Z-disc. Mutations in two genes, the β-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3), account for ~70 % of cases, >1,400 mutations have been identified thus far, mostly missense mutations [2].
Great phenotypic heterogeneity can be seen between and within families in terms of age at onset, degree and segmental distribution of hypertrophy, clinical manifestation, and outcome, suggesting a possible role for gene modifiers and environmental factors [2, 4]. The main four pathophysiological features and clinical manifestations of this CMP are LV diastolic dysfunction, LV outflow tract (OT) obstruction, heart failure (HF) symptoms, and sudden death (SD). The marked LV hypertrophy associated with increased ventricular wall stiffness due to interstitial fibrosis are responsible for LV diastolic dysfunction characterized by reduced compliance and abnormal relaxation, leading to diastolic HF. LV systolic function is usually preserved or is supernormal in the majority of patients, with a small subset (5–10 %) developing systolic dysfunction [e.g., LV ejection fraction (EF) <50 %) in the advanced stage (the so called end-stage evolution). This is a result of marked LV fibrosis, which is frequently associated with wall thinning and cavity dilatation. This unfavorable evolution, sometimes morphologically indistinguishable from a dilated cardiomyopathy, is associated with increased mortality (up to 11 % annual risk) and morbidity for HF and increased risk of SD [5, 6].
Dynamic LV OT obstruction can be appreciated in the resting condition in ~30 % of HCM patients (obstructive HCM), with a consequent dynamic LV OT pressure gradient at rest ≥30 mmHg. One third of patients show no LV OT obstruction at rest but only during exercise or other provocative maneuvers, such as Valsalva (labile LV OT obstruction). This peculiar feature of the disease is the consequence of vigorous systolic contraction, with an accelerated systolic ejection trough and narrowed LV OT due to systolic anterior motion (SAM) of the anterior mitral leaflet during mid systole, thus generating dynamic obstruction that assumes the aspect of a dynamic LV OT pressure gradient. The SAM of the mitral valve (MV), in association with abnormalities of the valve apparatus, leading to incomplete leaflet coaptation in mid systole, can also be responsible for a certain degree of mitral regurgitation (MR) that is often observed. SD in HCM seems to be less frequent than it was once thought. Recent data [2] indicate a prevalence of ~5 % in a hospital-based population. Nevertheless, HCM is the most common cause of SD in young athletes participating in competitive activity, without significant difference between genders [7]; SD may be the first clinical manifestation of the disease.
9.1.3 Clinical Diagnosis
As a result of its variability in phenotypic expression, the spectrum of clinical manifestations ranges from asymptomatic patients with normal life expectancy to severely symptomatic individuals. Dyspnea, angina pectoris under stress, palpitations, and occasionally syncope are the most common subjective manifestations, the most dramatic of which can be SD. SD most frequently occurs during or after strenuous physical exercise and is generally consequence of malignant arrhythmias.
Even if physical examination can often be silent, some affected patients manifest signs of systemic or pulmonary congestion, especially in the end stage of the disease. Irregular cardiac rhythm due to atrial fibrillation (AF) or presence of a fourth heart sound in patients in sinus rhythm can be detected on auscultation as a systolic murmur due to dynamic LV OT obstruction—the auscultatory stigmata of the disease—often associated with MR murmur. Every maneuver capable of reducing the preload or afterload (such as Valsalva or inhalation of amyl nitrite) or increasing contractility may result in an increase in intensity of this characteristic systolic heart murmur. Presence of particular syndromic findings (dysmorphic facies in patients with Noonan syndrome or typical maculopapular skin lesions in those with Anderson-Fabry disease) may suggest systemic disease presenting with an hypertrophied LV, which can be considered phenocopies of HCM [3, 8].
Electrocardiogram (ECG) is rarely normal in HCM patients, with about 6–10 % showing normal ECG at presentation [9]. More frequently, patients present with ECG signs of LV hypertrophy (Fig. 9.2), pathological Q waves, negative T waves, left-atrial (LA) enlargement, and intraventricular conduction abnormalities. Patients with a normal ECG at presentation seem to have a less advanced stage of the disease and a better prognosis [9].
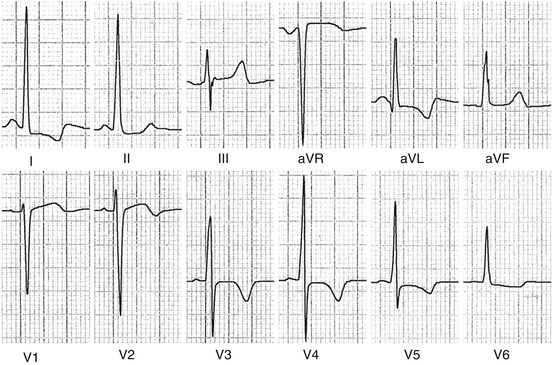
Fig. 9.2
Electrocardiograph (ECG) in a patient with hypertrophic cardiomyopathy (HCM) showing left ventricular (LV) hypertrophy and T-wave inversion
9.1.4 Other Diagnostic Tools
Ambulatory Holter ECG recording is part of the primary diagnostic and follow-up approach in detecting nonsustained ventricular tachycardia (e.g., an important risk factor for SD) and is therefore essential for risk stratification. Invasive electrophysiological examination has become less important in recent years [3]. The exercise test can be useful for risk stratification in selected patients for monitoring stress-induced ventricular arrhythmias and blood pressure response to physical exercise (systolic pressure fall) (see Chap. 13). Cardiac catheterization and endomyocardial biopsy are rarely employed in clinical practice.
9.1.5 Genetic Testing
Genetic testing is used to identify affected relatives in families known to have HCM. Because familial HCM is a dominant disorder, the risk that an affected patient will transmit the disease is 50 %. When a pathogenic mutation is identified in an index patient, genetic testing may be extended to each family member. Genetic counseling must be performed before genetic testing to improve patient and family understanding of implications of test results, potential risks, and benefits. Genetic counseling should be performed even if genetic testing is not undertaken. Genetic screening of first-degree relatives can identify family members carrying a mutation (genotype positive) but without overt cardiac hypertrophy (phenotype negative). In such cases, noninvasive clinical screening with physical examination, ECG, and 2D echocardiography or CMR is recommended due to the possibility of an age-related expression of the disease phenotype [1].
9.1.6 Natural History
HCM heterogeneity is manifested even in clinical presentation, symptoms severity, and clinical course (Table 9.1). Many affected patients (with the hypertrophic phenotype) present with no symptoms or major disability and experience a normal life expectancy without complications. On the other hand, symptomatic disease may present with different, not mutually exclusive, clinical settings. Some patients may experience SD, which can be the first and last manifestation of the disease, especially in individuals <35 years of age. Some other patients develop signs and symptoms of HF due to diastolic dysfunction (diastolic HF) with a well-preserved LV systolic function. A minority of those patients (~5–10 %) manifests progression toward end-stage disease, with impaired contractility and systolic dysfunction due to massive myocardial fibrosis. Those patients are at very high risk both for refractory HF and malignant arrhythmias. Finally, some affected individuals developed AF as a consequence of increased filling pressure and atrial dilatation, which can contribute to HF symptoms and brings with it major or minor systemic thromboembolic complications (e.g., cardioembolic stroke) [1, 10].
Table 9.1
Incidence of major cardiovascular events in HCM patients in the Heart Muscle Disease Registry of Trieste (1983–2013)