Abstract
Hypophosphatasia (HPP) results from mutations in the gene that encodes tissue nonspecific alkaline phosphatase ( TNSALP ). It can be inherited as both an autosomal recessive (AR) and autosomal dominant (AD) disorder. The forms are named for age at onset of symptomatology and include the perinatal (lethal; AR), infantile (AR), childhood (AR and AD), and adult (AD) hypophosphatasia. In many of the cases that are diagnosed in the early perinatal period, infants are either stillborn or die in the immediate neonatal period from respiratory insufficiency owing to abnormal thorax and pulmonary hypoplasia. Perinatal hypophosphatasia should be suspected if prenatal ultrasound reveals poor mineralization of the cranium, ribs, and appendicular skeleton. The long bones are thin and short, and they can be bowed with lateral spikes at the metaphyseal ends. It is important to differentiate this disorder from osteogenesis imperfecta, and post-delivery radiographs can be diagnostic.
Keywords
hypophosphatasia, HPP, TNSALP , Bowdler spur, hypomineralization
Introduction
Hypophosphatasia (HPP) is an autosomal recessive disorder that is characterized by poor mineralization of bones and teeth. There are at least six phenotypes, which are distinguished from each other by age at diagnosis, inheritance pattern, and prognosis. The primary forms are named for age at diagnosis and include perinatal (lethal), infantile, childhood, and adult HPP. In most cases that are diagnosed perinatally, infants are either stillborn or die in the immediate neonatal period from respiratory insufficiency owing to abnormal thorax and pulmonary hypoplasia, although there are cases with nonlethal outcomes. Infantile HPP manifests by 6 months of age and is associated with failure to thrive, premature craniosynostosis, respiratory complications from rickets of the chest, and seizures. Childhood HPP manifests with early loss of deciduous teeth and bone pain. The first sign of the adult form of HPP is often foot pain from stress fractures.
Other, very rare forms are odontohypophosphatasia and pseudohypophosphatasia; the former usually manifests in middle age with loss of adult teeth, and is associated with only dental and biochemical abnormalities, and the latter clinically and radiologically appears the same as the infantile form except serum alkaline phosphatase is normal. This chapter focuses on the perinatal subtype because the diagnosis can be suggested by prenatal ultrasound (US).
Disorder
Definition
HPP is a disorder of bone mineralization leading to skeletal abnormalities (osteomalacia, rickets). The clinical presentation ranges from perinatal lethality to a relatively benign course in which symptoms (premature loss of teeth or fractures, or both) first manifest in adulthood.
Prevalence and Epidemiology
The incidence of lethal HPP is estimated at 1 : 100,000 births. In the inbred population of the Canadian Mennonites, the incidence is increased to 4 : 10,000 live births based on a founder effect.
Etiology, Pathophysiology, and Embryology
Severe forms (perinatal, infantile) of HPP are inherited in an autosomal recessive fashion. Inheritance of the milder forms (childhood, adult, odontohypophosphatasia) is less clear; there are reports of both autosomal dominant and autosomal recessive patterns. The perinatal form of HPP is caused by a deficiency of tissue-nonspecific alkaline phosphatase (TNSALP) in osteoblasts and chondrocytes, resulting in reduced levels of serum alkaline phosphatase. Bone mineralization is impaired, leading to osteomalacia, and perinatal death occurs as a result of pulmonary hypoplasia secondary to small chest size. Carriers can be recognized by a low level of serum alkaline phosphatase and increased urinary phosphoethanolamine levels.
Manifestations of Disease
Clinical Presentation
The diagnosis of perinatal HPP is suspected on prenatal US, but molecular genetics or radiologic evaluation, or both, are required for definitive diagnosis. Many affected fetuses are stillborn. Infants who survive have short limbs; poor skeletal mineralization (especially of the spine); bowed lower extremities; and bone spurs on the long bones, knees, and elbows. Death is usually from respiratory failure owing to the small chest and subsequent hypoplastic lungs. Fever of unknown origin, anemia, hypercalcemia, seizures, and intracranial hemorrhage may also be seen. The diagnosis is confirmed by a decrease in serum alkaline phosphatase activity, molecular genetics, serum TNSALP activity, and radiologic findings.
Imaging Technique and Findings
Ultrasound.
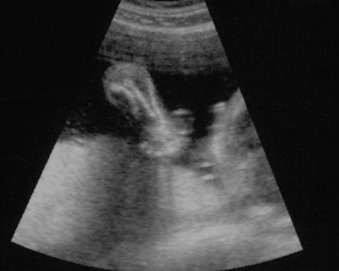
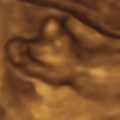
Perinatal HPP should be suspected if prenatal US reveals poor mineralization of the cranium, ribs, and thoracic spine. Craniosynostosis may be present, though this is rare. Lack of mineralization of the hand can be a very helpful diagnostic clue ( Fig. 51.1 ). Poor mineralization of the calvarium ( Fig. 51.2 ), short and thin long bones, bowed lower extremities, and cup-shaped metaphyses (lateral spikes) ( Fig. 51.3 ) further specify the diagnosis. Bony spurs are often present but may be difficult to see by two-dimensional US; three-dimensional US may improve visualization ( Fig. 51.4 ). A thickened nuchal translucency in the first trimester may be apparent, and polyhydramnios may occur in later trimesters. Three-dimensional or four-dimensional US is useful in the evaluation of fetuses with skeletal dysplasias, especially for imaging the face and extremities.