Jyoti Kumar, Abhinav C. Bhagat The intricate anatomy and specialization of the ear as a hearing and balancing organ lends a unique nature to it. The different origins and development of conducting and neurosensory apparatus are responsible for a diverse set of abnormalities afflicting the ear. In this chapter, we limit ourselves to discussion of inner ear, focussing on brief embryology, normal imaging anatomy, imaging techniques, classification and imaging features of congenital and acquired pathologies of inner ear. The development of inner ear occurs in a precise step-by-step manner starting third week of gestation. It follows that maturation arrest at different developmental stages results in different malformations. The lateral surface of neuroectoderm develops a plate-like thickening in the third week, called the otic placode. Invagination of the otic placode occurs in the fourth week to form an epithelium-lined fluid-containing cavity known as the otic vesicle or otocyst. The otocyst buries into the head mesenchyme which surrounds it. The mesenchyme around the otocyst forms the cartilaginous otic capsule. The otocyst is the primordial membranous labyrinth which undergoes further differentiation, giving rise to cochlear, vestibular and endolymphatic sac appendages. The cochlear duct develops over the next 3 weeks and gains 11/2 turn during the eighth week and 21/2 turns at the end of 10th week. The vestibule and semicircular canals are completely developed by the 11th week and 19th–22nd weeks, respectively. The superior semicircular canal is the first to form followed by posterior and lateral semicircular canals. The cartilaginous otic capsule undergoes partial vacuolization to form perilymphatic spaces and undergoes ossification between the 16th and 23rd weeks. The endolymphatic duct and sac attain their adult size after puberty. In recent years, there has been an increase in our understanding of molecular and genetic mechanisms contributing to inner ear development. Specific genetic mutations responsible for characteristic inner ear malformations have been identified. Additionally, the genes responsible for inner ear development are also involved in development of other organ systems. These findings explain why inner ear malformations have syndromic associations. The inner ear anatomy is discussed in detail in Chapter 3 Imaging technique, anatomy and embryology; few other salient features are mentioned here. The variety of tissues in the temporal bone facilitates imaging evaluation by both high-resolution computed tomography (HRCT) and magnetic resonance (MR) imaging. Both these modalities have their utility in comprehensive evaluation of temporal bone, such that neither one can be considered superior or inferior to the other. While CT performs well in assessment of air spaces and osseous structures, MR imaging is indispensable for evaluation of membranous labyrinth, neural elements and brain parenchyma. Therefore, both these modalities can be used in a complementary manner for identification of pathology. HRCT of the temporal bone using a multidetector CT scanner (64 slices or more) provides good anatomic details for the evaluation of inner ear. It also gives useful information regarding status of middle and external ear. Scan plane: Axial scans are acquired (512 × 512 matrix) from the top of petrous apex to inferior tip of mastoid bone using spiral technique. Each ear is thereafter reconstructed on a smaller FOV ranging between 80 and 100 mm. Major disadvantages include poor soft-tissue resolution in bony labyrinth and IAC. Radiation exposure is another drawback. 3T MR imaging system provides excellent images for inner ear examination. Lack of widespread availability and need for sedation in very young children are some of the disadvantages. The spectrum of congenital inner ear anomalies is broad and consequently so is the number of various classifications which have been proposed over time. Initially, these malformations were classified on the basis of histopathologic findings. Such classifications incorporated Bing–Siebenmann (BS), Scheibe, Mondini, Alexander and Michel-type dysplasias. In 1987, Jackler et al. proposed a classification system based on the radiological appearance of these malformations. They built on the hypothesis that maturation arrest at different stages of inner ear embryogenesis results in various malformations. The congenital bony cochlear malformations were categorized into five types: They had stated that the term “Mondini dysplasia” was being used to describe virtually any congenital malformation of the bony labyrinth detectable on radiographic examination. With their classification system, they were successful in avoiding the misuse of the term “Mondini dysplasia”. The malformations involving other inner ear structures with normal cochlea were classified into two types by Jackler et al: vestibule–lateral semicircular canal dysplasia (enlarged vestibule with a short, dilated lateral semicircular canal and normal remaining semicircular canals); and enlarged vestibular aqueduct (accompanied by normal semicircular canals and normal or enlarged vestibule). Sennaroglu and Saatci proposed a new system for classification of inner ear malformations in 2002, wherein they went a step ahead from the classification proposed by Jackler et al. and resolved the dilemma in the diagnosis of Mondini deformity. In the process of evaluating inner ear malformations using HRCT, they stated that the term “Mondini deformity” was being used to describe two different types of IP of the cochlea. The first condition consisted of a completely empty cystic appearing cochlea with absence of the cribriform area and entire modiolus, accompanied by a cystic vestibule. The second condition comprised 1.5 turns of cochlea with confluence of middle and apical turns into a cystic cavity, dilated vestibule and enlarged vestibular aqueduct, consistent with the classic description of Mondini deformity. Therefore, they classified these malformations (in descending order of severity) as follows: Michel deformity, cochlear aplasia, common cavity, IP-I (cystic cochleovestibular malformation), CH and IP-II (Mondini deformity), which according to them were clinically more useful. In 2017, Sennaroglu and Bajin modified their previous classification and divided the inner ear malformations into eight distinct groups. This is the latest classification, and the malformations described in this chapter will be described using the same. As per the latest classification by Sennaroglu and Bajin, inner ear malformations are currently classified into eight types. The clinical features, imaging findings, imaging differentials and treatment options for each of these are described in the following section. The diversity of congenital malformations gives rise to varied clinical manifestations. The most common presentation is with hearing and speech impairment. These patients can be congenitally symptomatic or may present in childhood or even later. Most common presentation is with variable degree of sensorineural hearing loss that depends on the severity of the underlying malformation. Mixed conductive and sensorineural hearing loss or sometimes even pure conductive hearing loss may also occur when there is accompanying stapedial fixation. The malformations may also occur as a component of syndromes, which can significantly impact management. Additionally, these patients may be predisposed to serious complications such as recurrent meningitis. Inner ear involvement is known to occur in various syndromes. Inner ear manifestations of CHARGE (coloboma, heart defects, choanal atresia, retarded growth, genital abnormalities and ear abnormalities) syndrome include CH and cochlear aperture stenosis in association with absent semicircular canals. Branchio-oto-renal syndrome is characterized by CH (offset of the hypoplastic middle and apical turns away from the basal turn), early exit of labyrinthine segment of the facial nerve from the IAC and ossicular chain dysplasia. Other systemic manifestations include cervical branchial apparatus anomalies and renal anomalies. In Waardenburg syndrome, there is absence of posterior semicircular canal with slightly dilated lateral semicircular canal and CH type 3. Other manifestations include pigmentation abnormalities (white forelock, hypopigmented skin patches, heterochromia irides), hypertelorism, cerebellar dysplasia and Hirschsprung’s disease, among others. Down syndrome is associated with CH, cochlear aperture stenosis and dilated cochlear aqueduct with characteristic facies. Patients with Pendred syndrome have a high incidence of Mondini malformation, apart from thyroid involvement from goitre. It is a rare anomaly at the most severe end of the spectrum of inner ear malformations. Known by the eponym of Michel aplasia, it occurs due to failure of development of otic placode during the third week of gestation. Apart from inner ear absence, it is associated with abnormal development of derivatives of second branchial arch (absence of stapes, abnormal course of facial nerve) and skull base abnormalities (platybasia, aberrant course of internal jugular vein). Imaging findings
3.33: Imaging inner ear
Introduction
Embryology
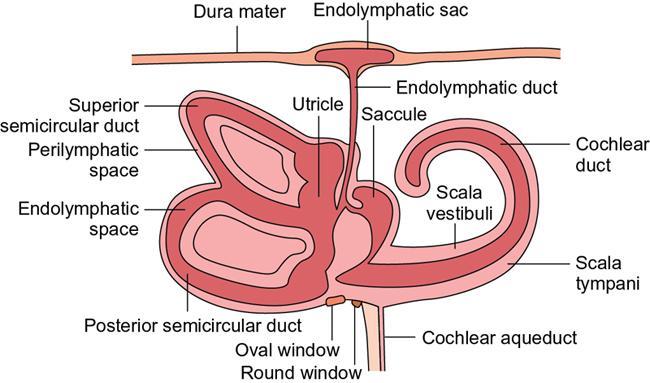
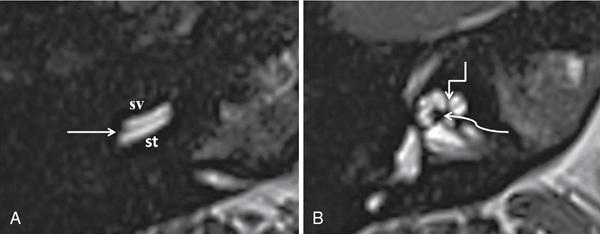
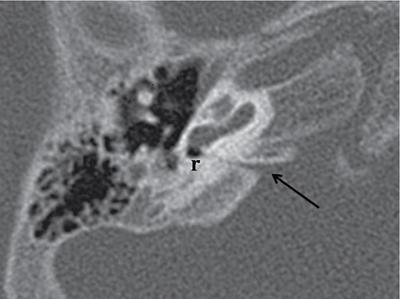
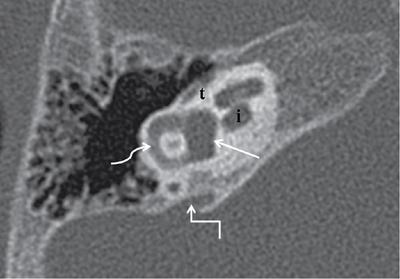
Normal imaging anatomy
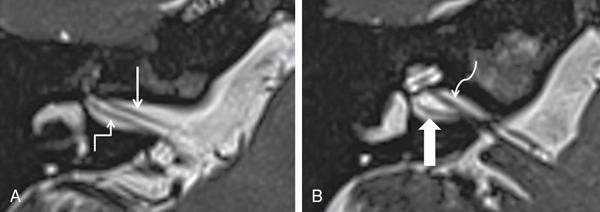
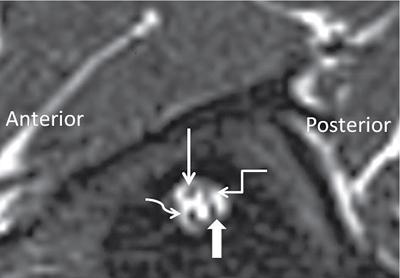
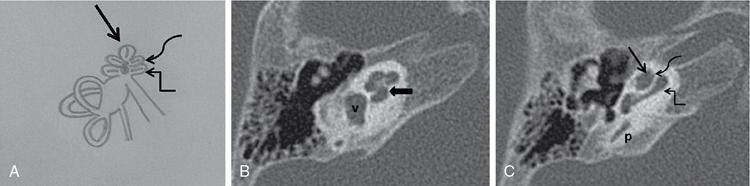
Imaging techniques
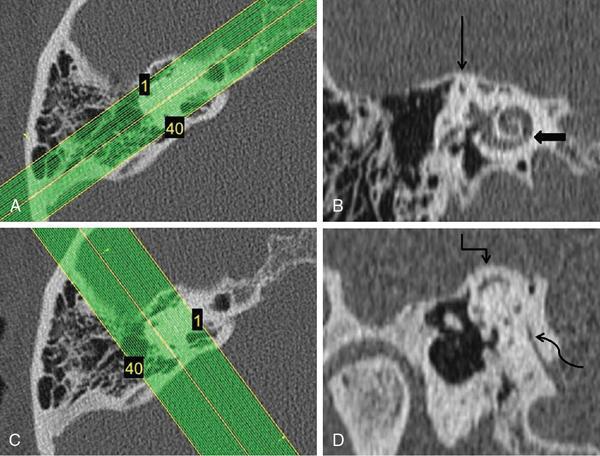
Computed tomography protocol
Magnetic resonance imaging protocol
Classification of congenital inner ear malformations
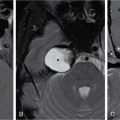
Congenital anomalies of inner ear
Clinical features
Inner ear anomalies
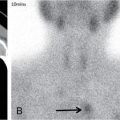
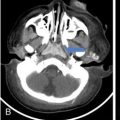
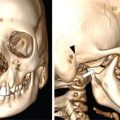
Complete labyrinthine aplasia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


