LEARNING OBJECTIVES
1. List and identify on a chest radiograph and computed tomographic (CT) scan the four patterns of interstitial lung disease (ILD): linear, reticular, reticulonodular, and nodular.
2. Make a specific diagnosis of ILD when supportive findings are present in the history or on radiologic imaging (e.g., dilated esophagus and ILD in scleroderma; enlarged heart, pacemaker or defibrillator, prior sternotomy, and ILD in a patient with amiodarone drug toxicity).
3. Identify Kerley A and B lines on a chest radiograph and CT scan, and explain their etiology and significance.
4. Recognize the changes of congestive heart failure on a chest radiograph (enlarged cardiac silhouette, pleural effusions, vascular redistribution, interstitial or alveolar edema, Kerley lines, enlarged azygos vein, increased ratio of artery-to-bronchus diameter).
5. Define “asbestos-related pleural disease” and “asbestosis”; identify each on a chest radiograph and CT scan.
6. Describe what a “B reader” is, as related to the evaluation of pneumoconioses.
7. Identify honeycombing on a chest radiograph and CT scan, state the significance of this finding (end-stage lung disease), and list the common causes of honeycomb lung.
8. Recognize progressive massive fibrosis/conglomerate masses secondary to silicosis or coal worker’s pneumoconiosis on a chest radiograph and CT scan.
9. Recognize the typical appearance and upper lobe–predominant distribution of irregular lung cysts or nodules on chest CT of a patient with Langerhans cell histiocytosis (LCH).
10. List four causes of unilateral ILD (aspiration, radiation, lymphangitic carcinomatosis secondary to lung cancer, asymmetric edema).
11. List the common causes of lower lobe–predominant ILD (idiopathic pulmonary fibrosis, asbestosis, chronic aspiration, collagen vascular disease).
12. List two causes of upper lobe–predominant ILD (chronic hypersensitivity pneumonitis, sarcoidosis).
13. Recognize the findings of lymphangioleiomyomatosis (LAM) on a chest radiograph and CT scan.
This chapter on interstitial lung disease (ILD) is followed by a chapter on alveolar lung disease (ALD). When the chest radiograph shows a clear pattern of ILD or ALD, one can render a differential diagnosis on the basis of the pattern of parenchymal disease (Table 3.1). A conundrum arises when widespread small opacities are difficult to categorize into one group or the other on chest radiography, or when ILD and ALD are both present. In these cases, coming up with a differential diagnosis is not as straightforward. One must decide what the predominant pattern is, take into consideration the clinical history and any associated radiographic findings, or further define the pattern(s) and distribution of disease with a CT scan of the lungs.
Diffuse lung disease is a term often used to describe a group of disorders of known cause (e.g., collagen vascular disease, environmental or drug-related) as well as disorders of unknown cause. The latter include idiopathic interstitial pneumonias, granulomatous lung disorders (e.g., sarcoidosis), and other forms of ILD including lymphangioleiomyomatosis (LAM), Langerhans cell histiocytosis (LCH), and eosinophilic pneumonia (1). The diagnostic process in diffuse lung disease begins with a clinical evaluation that includes a history, physical examination, chest radiograph, and lung function tests. Many patients will then undergo CT scanning of the chest, and, depending on the findings, may proceed to transbronchial biopsy, bronchoalveolar lavage, or surgical lung biopsy.
PATTERNS OF ILD
The interstitium of the lung is not normally visible radiographically; it becomes visible only when disease (e.g., edema, fibrosis, tumor) increases its volume and attenuation. The interstitial space is defined as “a continuum of loose connective tissue throughout the lung composed of three subdivisions: (i) the bronchovascular (axial), surrounding the bronchi, arteries, and veins from the lung root to the level of the respiratory bronchiole; (ii) the parenchymal (acinar), situated between the alveolar and capillary basement membranes; and (iii) the subpleural, situated beneath the pleura, as well as in the interlobular septa” (2). Any or all of these three interstitial compartments can be abnormal at any one time.
Table 3.1 DIFFERENTIAL DIAGNOSIS OF INTERSTITIAL LUNG DISEASE
“BADLASH”
Bronchiectasis (ILD “look-alike”)
Bugs (especially fungi, Mycoplasma, and viruses)
Aspiration, chronic
Amyloidosis
Drug toxicity
Lymphangioleiomyomatosis
Lymphangitic carcinomatosis
Lymphoma
Lymphoid interstitial pneumonia and other idiopathic interstitial pneumonias
Asbestosis
Sarcoidosis
Scleroderma and other collagen vascular diseases
Silicosis
Hypersensitivity pneumonitis
Heart failure
Histiocytosis (Langerhans cell histiocytosis)
ILD, interstitial lung disease
ILD may result in four patterns of abnormal opacity on chest radiographs and CT scans: linear, reticular, nodular, and reticulonodular (Fig. 3.1). These patterns are more accurately and specifically defined on CT. A linear pattern is seen when there is thickening of the interlobular septa, producing Kerley lines. These septal lines were first described by Kerley in patients with pulmonary edema (3). Kerley B lines are short, straight lines (1 to 2 cm) perpendicular to and abutting the lower lateral pleural edge. Kerley A lines are generally longer (2 to 6 cm), they radiate out from the hilum toward the pleura but are not contiguous with the pleura, and they are most obvious in the upper and middle lungs. The interlobular septa contain pulmonary veins and lymphatics. The most common cause of interlobular septal thickening, producing Kerley A and B lines, is pulmonary edema, as a result of pulmonary venous hypertension and distension of the lymphatics (Figs. 3.2 and 3.3). Other causes of Kerley lines are listed in Table 3.2. Anything that causes thickening of the interlobular septa can produce Kerley lines, including edema, inflammation, tumor, or fibrosis. Septal thickening without architectural distortion is more likely to represent pulmonary edema.
A reticular pattern results from the summation or superimposition of irregular linear opacities. The term reticular is defined as meshed, or in the form of a network. Reticular opacities can be described as fine, medium, or coarse, as the width of the opacities increases. A classic reticular pattern is seen with pulmonary fibrosis, in which multiple curvilinear opacities form small cystic spaces along the pleural margins and lung bases (honeycomb lung) (Fig. 3.4).
A nodular pattern consists of multiple round opacities, generally ranging in diameter from 1 mm to 1 cm, which may be difficult to distinguish from one another as individual nodules on a chest radiograph. Nodular opacities may be described as miliary (1 to 2 mm, the size of millet seeds), small, medium, or large, as the diameter of the opacities increases (Figs. 3.5 and 3.6). A nodular pattern, especially with an upper lung–predominant distribution, suggests a specific differential diagnosis (Table 3.3; Figs. 3.7 and 3.8).
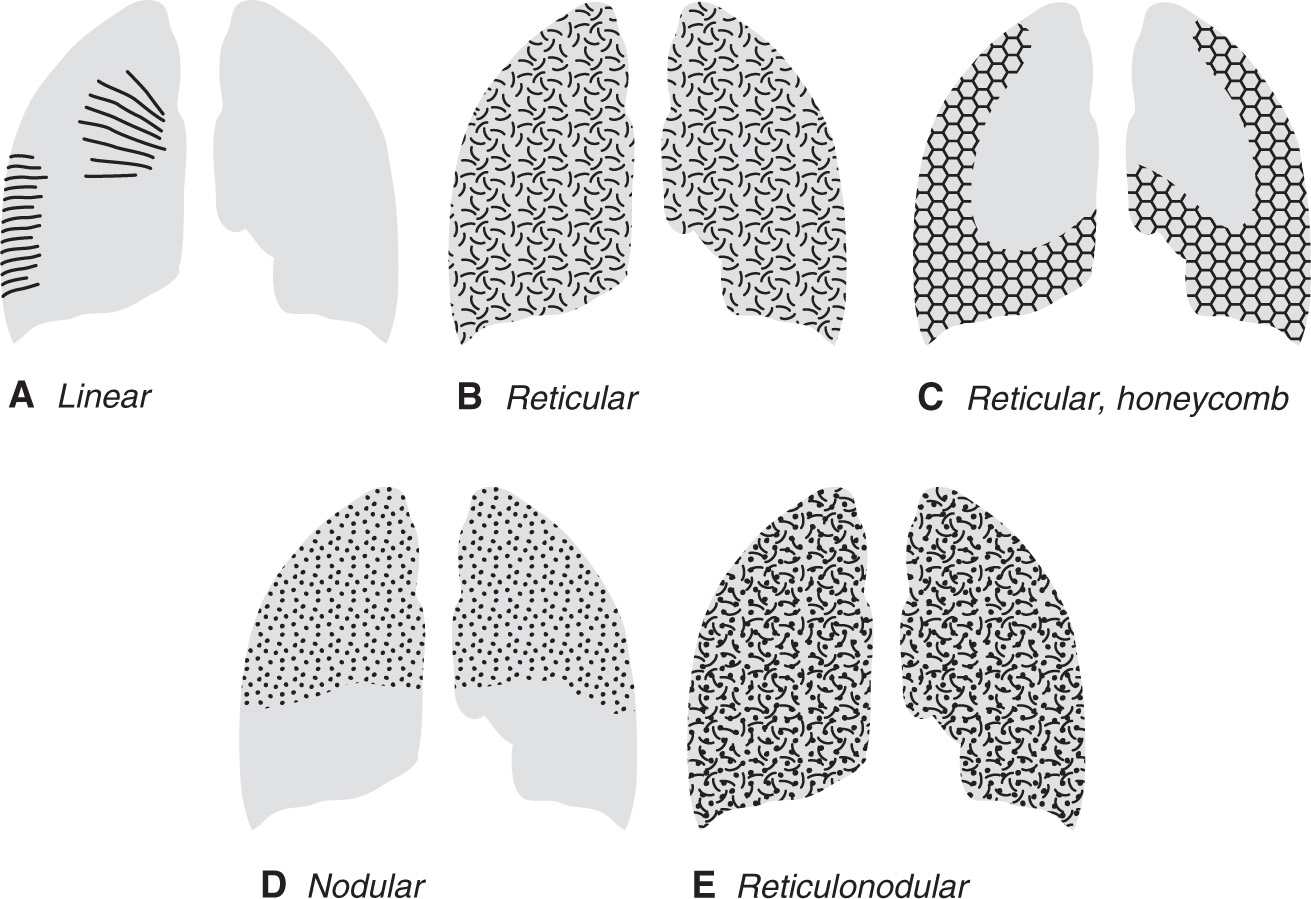
FIG. 3.1 • Diagrams illustrating the four types of ILD. A: Linear ILD is seen as Kerley lines. Kerley A lines radiate out from the hila to the periphery of the lung. Kerley B lines are shorter lines that contact and are perpendicular to the lateral pleural edge, predominantly in the lower lungs. Both A and B lines are seen as a result of interlobular septal thickening, most commonly from pulmonary edema. B: Reticular ILD is seen as a network of curvilinear opacities. When seen as a result of a reversible process, such as viral pneumonia, sarcoidosis, or hypersensitivity pneumonitis, the distribution can be patchy or diffuse. C: When reticular ILD is seen as a result of chronic, irreversible lung disease, such as usual interstitial pneumonia, honeycombing is seen. The curvilinear opacities form small cystic spaces (forming the honeycomb) in a characteristic bibasilar and subpleural distribution. D: Nodular ILD will often, but not always, have an upper and middle lung–predominant distribution. This is often the case with sarcoidosis, LCH, silicosis, and coal worker’s lung. The nodules generally range from 1 to 10 mm in size. E: Reticulonodular ILD results from a combination of reticular and nodular opacities, or it can be caused by reticular opacities seen end-on. This pattern is often difficult to distinguish from a pure nodular or reticular pattern on chest radiography. The list of diagnostic possibilities to consider when this pattern is seen can be shortened by taking into account the acuity of the disease, the distribution of disease, and associated radiographic abnormalities.
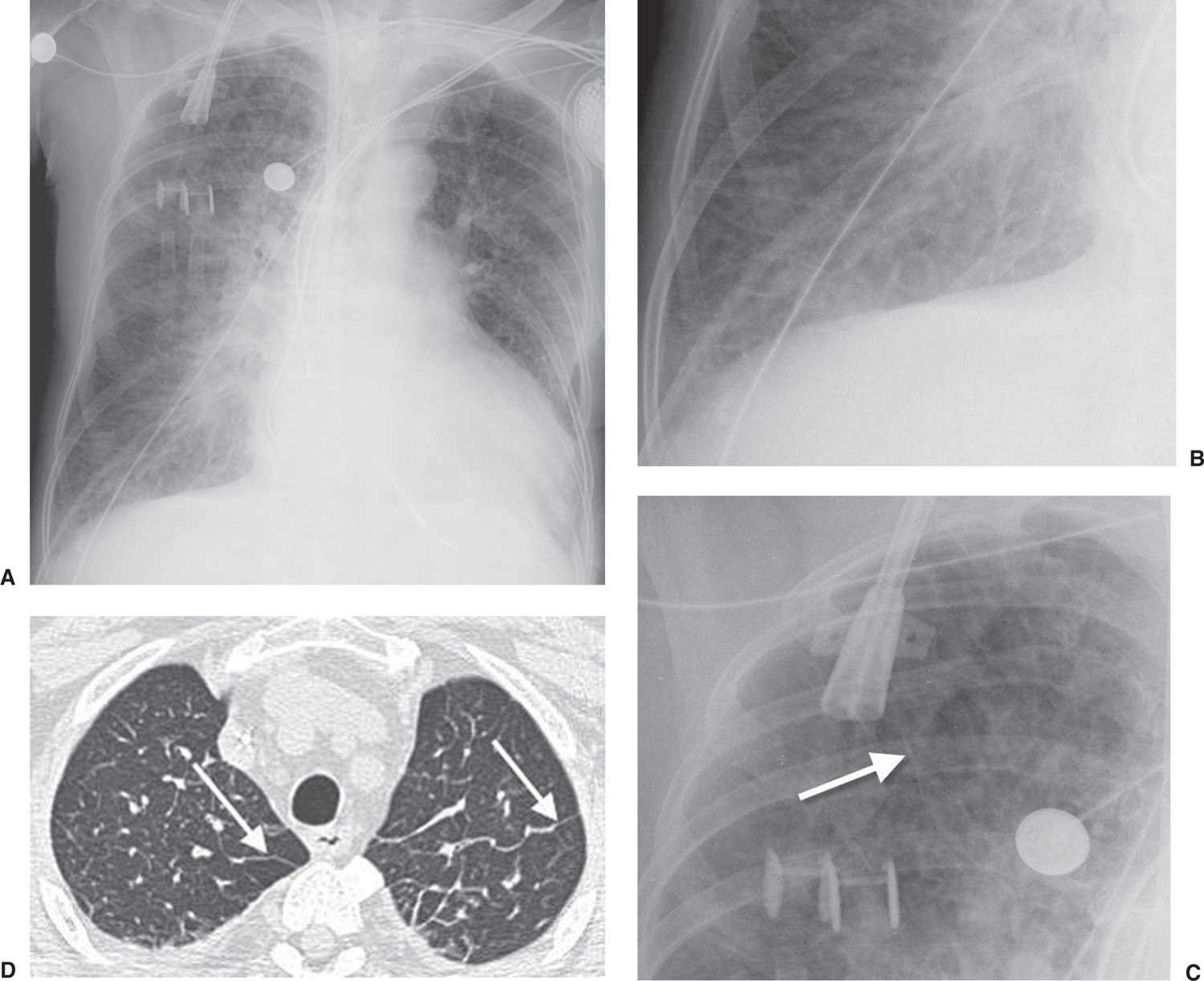
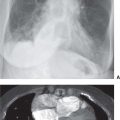
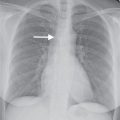
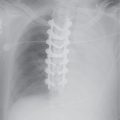
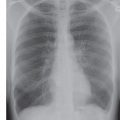
FIG. 3.2 • Cardiogenic edema and Kerley lines. A: PA chest radiograph shows an enlarged cardiac silhouette and bilateral reticular and linear ILD. B: Close-up view of (A), lower right lung, shows short, linear opacities perpendicular to the lateral pleural edge, representing Kerley B lines. C: Close-up of (A), right upper lung, shows linear opacities (arrow) radiating outward from the hila, representing Kerley A lines. D: CT shows interlobular septal thickening (arrows), representing Kerley lines.
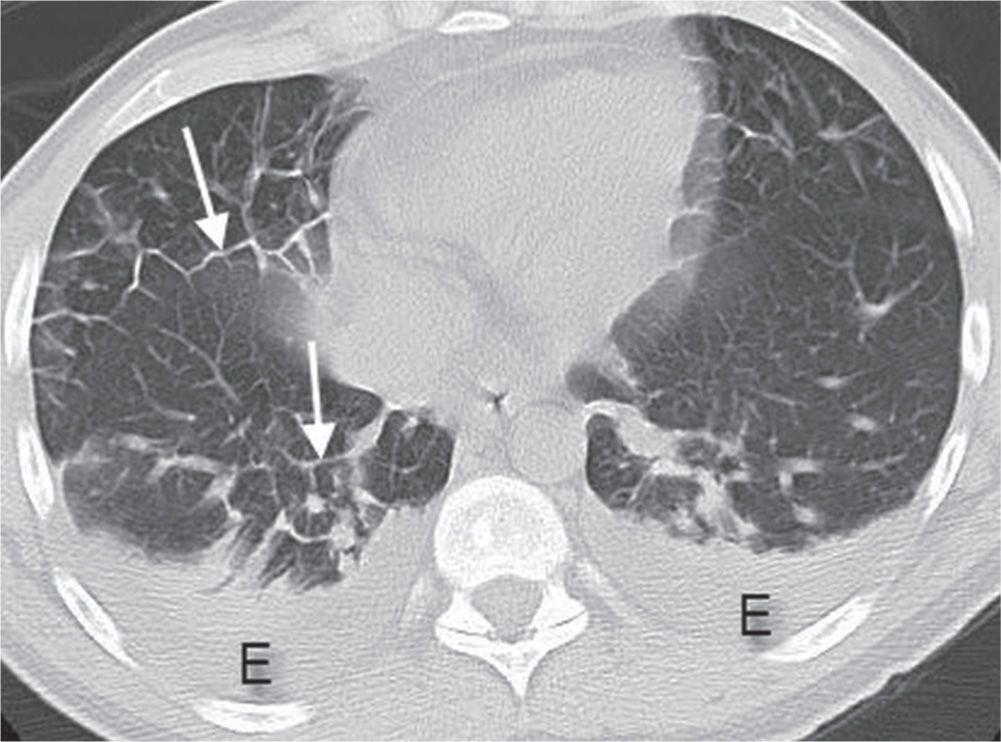
FIG. 3.3 • Cardiogenic edema and Kerley lines. CT scan shows septal thickening (Kerley lines, arrows), small areas of ground-glass opacity, and bilateral pleural effusions (E).
Table 3.2 DIFFERENTIAL DIAGNOSIS OF KERLEY LINES
Pulmonary edema—the most common cause
Mitral stenosis
Lymphangitic carcinomatosis
Malignant lymphoma
Congenital lymphangiectasia
Viral and Mycoplasma pneumonias
Idiopathic pulmonary fibrosis
Pneumoconiosis
Sarcoidosis
Late-stage hemosiderosis
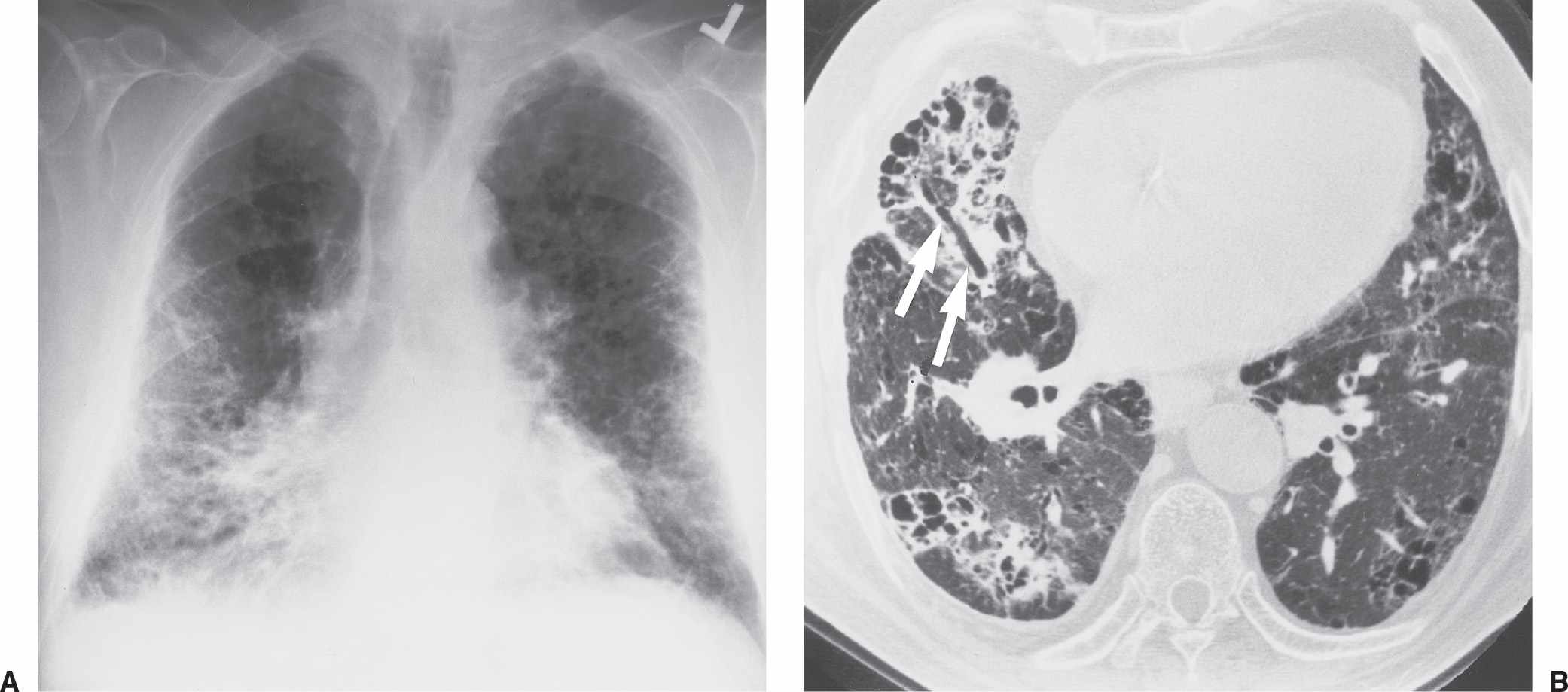
FIG. 3.4 • Farmer’s lung and pulmonary fibrosis. This 50-year-old man presented with end-stage lung fibrosis from chronic exposure to inhaled antigens on his farm. A: PA chest radiograph shows medium to coarse reticular ILD with a predominant bibasilar and subpleural distribution. B: CT scan shows multiple small cysts (honeycombing) involving predominantly the subpleural peripheral regions of lung. Traction bronchiectasis, another sign of end-stage lung fibrosis, is seen in the right middle lobe (arrows).
A reticulonodular pattern results from a combination of reticular and nodular opacities, or it can appear when reticular opacities are seen end-on. This pattern is often difficult to distinguish from a purely reticular or nodular pattern, and in such a case a differential diagnosis should be developed on the basis of the predominant pattern. If there is no predominant pattern, causes of both nodular and reticular patterns should be considered. An acute appearance suggests pulmonary edema or pneumonia (Figs. 3.9 and 3.10). A lower lung–predominant distribution with decreased lung volumes suggests idiopathic pulmonary fibrosis, asbestosis, collagen vascular disease, or chronic aspiration. A reticulonodular pattern and larger-than-normal lung volumes can be seen with LAM and LCH. A middle or upper lung–predominant distribution suggests mycobacterial or fungal disease, silicosis, sarcoidosis, LCH, extrinsic allergic alveolitis (hypersensitivity pneumonitis), or, very rarely, ankylosing spondylitis. Kerley lines help limit the differential diagnosis (see Table 3.2). Associated lymphadenopathy suggests sarcoidosis; neoplasm (lymphangitic carcinomatosis, lymphoma, metastases); infection (viral, mycobacterial, or fungal); and silicosis. Associated pleural thickening and/or calcification suggest asbestosis. Associated pleural effusion suggests pulmonary edema, lymphangitic carcinomatosis, lymphoma, collagen vascular disease, or LAM (especially if the effusion is chylous). Associated pneumothorax suggests LAM or LCH.
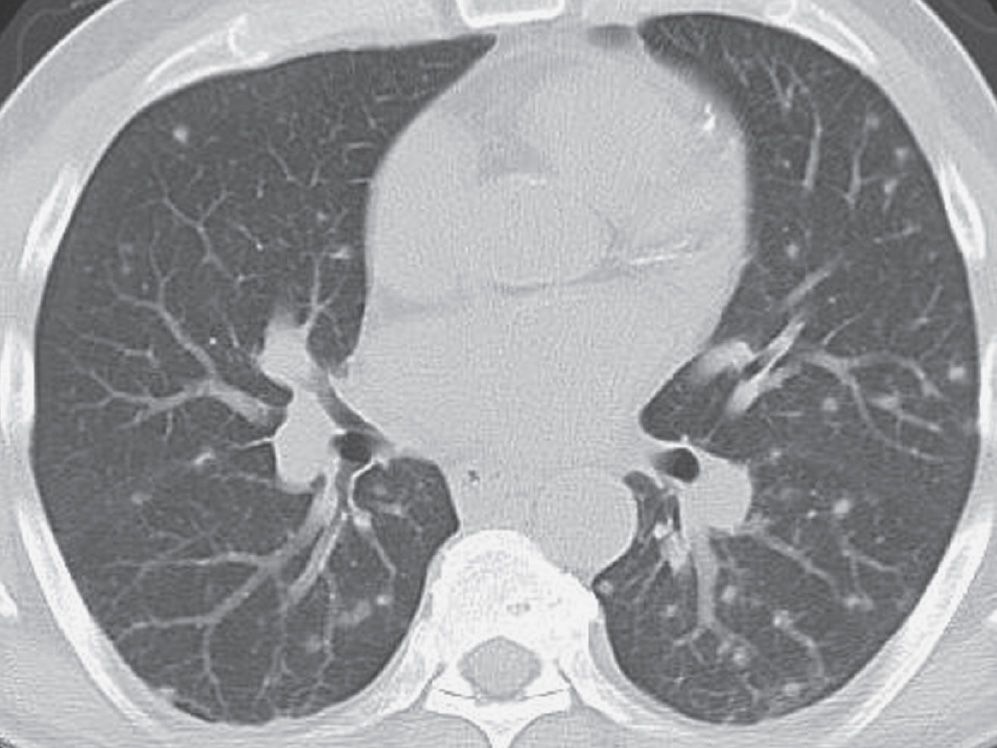
FIG. 3.5 • Disseminated histoplasmosis and nodular ILD. This previously healthy man living in the upper midwestern part of the United States presented with mild symptoms of shortness of breath and cough. CT scan shows multiple bilateral, round, pulmonary nodules.
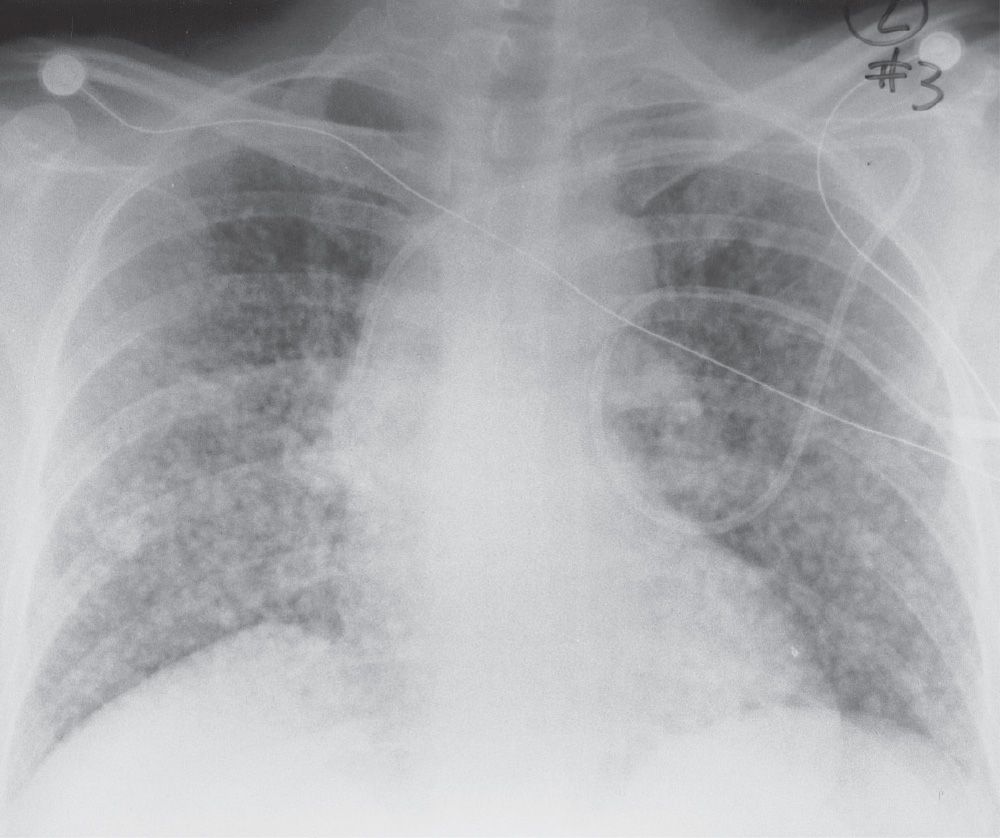
FIG. 3.6 • Hematogenous metastases and nodular ILD. This 45-year-old woman presented with metastatic gastric carcinoma. The PA chest radiograph shows a diffuse pattern of nodules, 6 to 10 mm in diameter.
Table 3.3 DIFFERENTIAL DIAGNOSIS OF A NODULAR PATTERN OF INTERSTITIAL LUNG DISEASE
“SHRIMP”
Sarcoidosis
Histiocytosis (Langerhans cell histiocytosis)
Hypersensitivity pneumonitis
Rheumatoid nodules
Infection (mycobacterial, fungal, viral)
Metastases
Microlithiasis, alveolar
Pneumoconioses (silicosis, coal worker’s, berylliosis)
This list excludes the relatively uncommon diagnosis of amyloidosis.
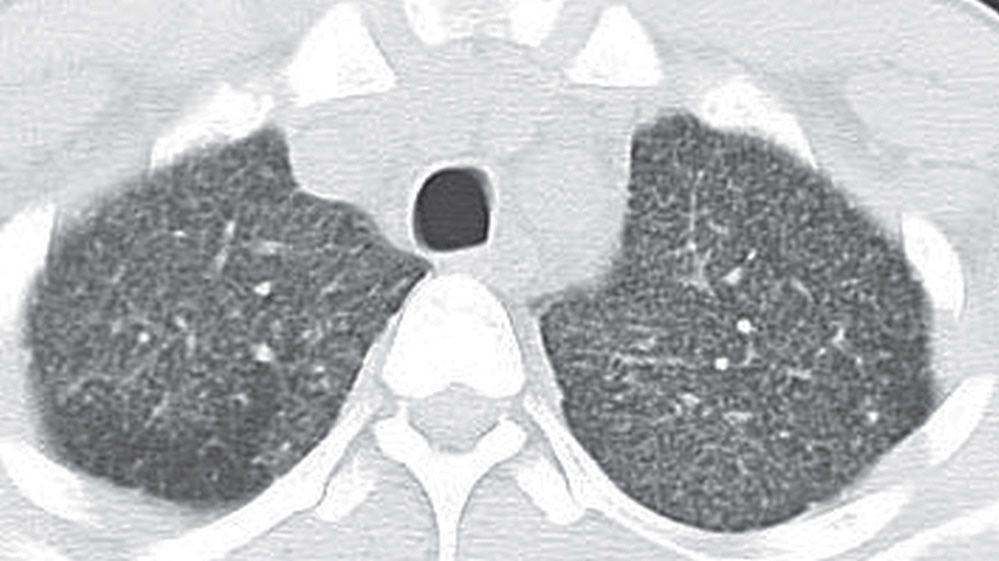
FIG. 3.7 • Miliary tuberculosis and nodular ILD. This 3-year-old Hispanic boy with acquired immunodeficiency syndrome presented with a 2-week history of cough, fever, chills, night sweats, and headache. CT scan shows numerous tiny nodules in a random distribution.
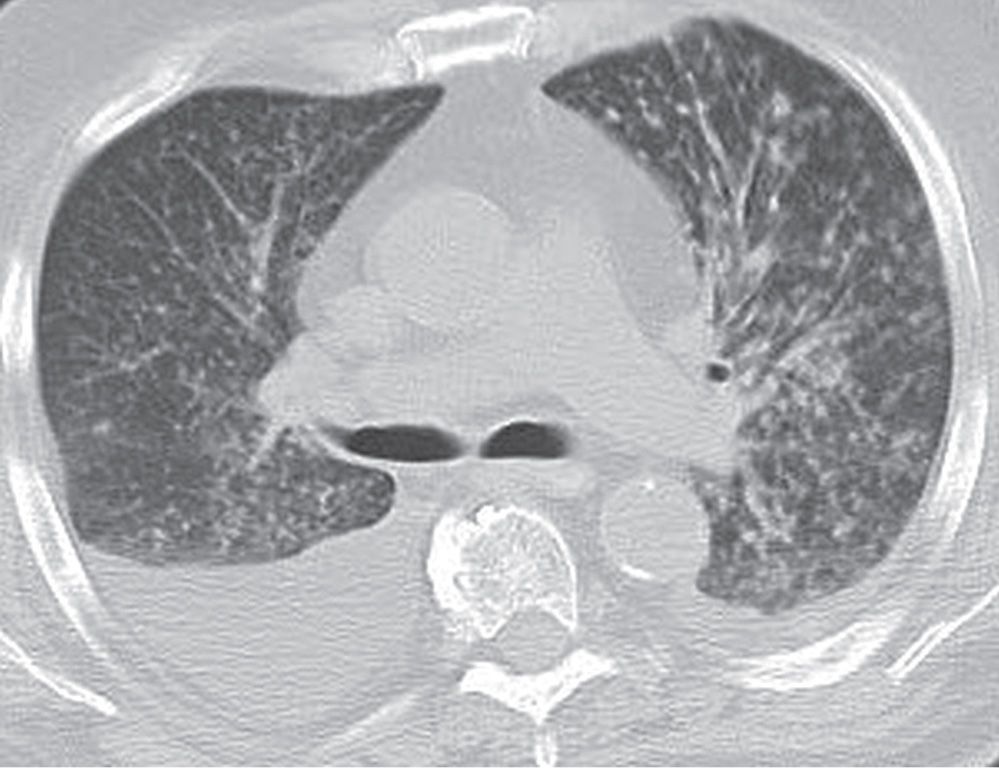
FIG. 3.8 • Coccidioidomycosis and nodular ILD. CT scan of a patient living in Arizona shows numerous small nodules in a random distribution and pleural effusions.
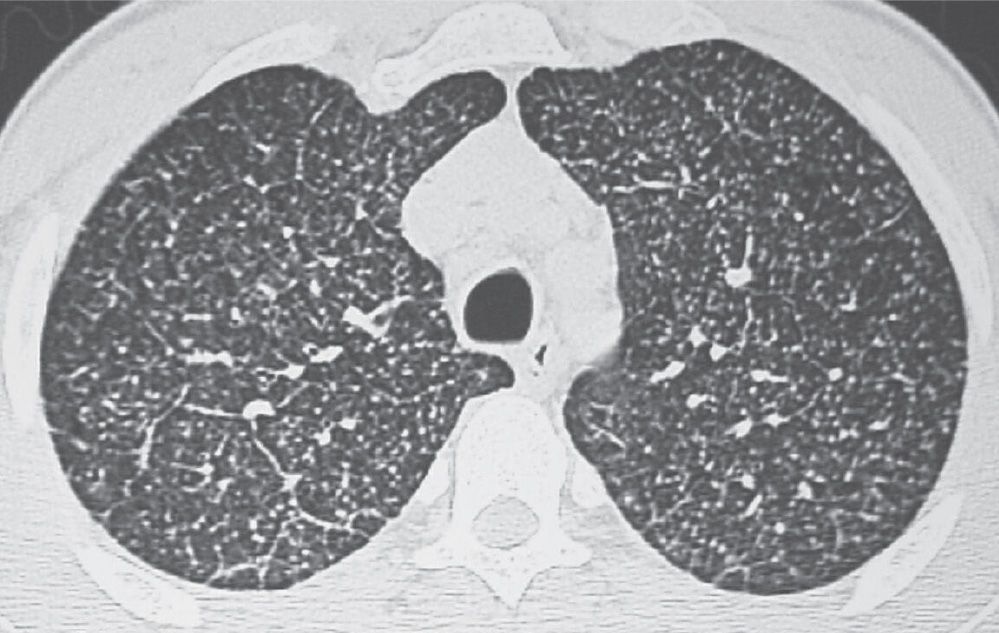
FIG. 3.9 • Disseminated histoplasmosis and reticulonodular ILD. CT scan shows multiple circumscribed, round pulmonary nodules, 2 to 3 mm in diameter, and scattered reticular opacities.
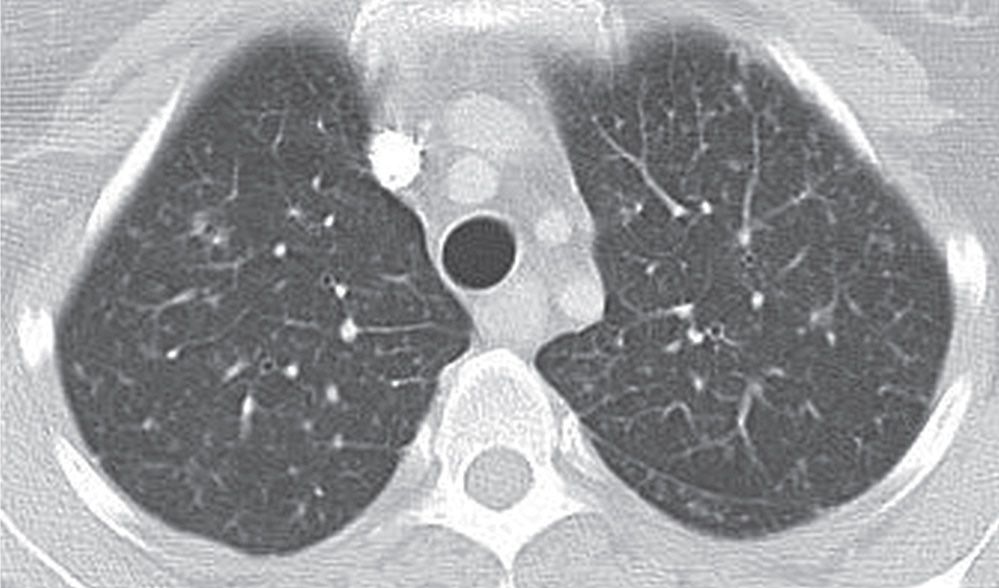
FIG. 3.10 • Aspergillosis and reticulonodular ILD. CT scan shows small nodules, reticular opacities, and septal thickening.
PULMONARY EDEMA
Hydrostatic pulmonary edema is defined as abnormal water in the lungs secondary to elevated pulmonary venous pressure from a failing left ventricle, mitral stenosis, increased circulating blood volume (as with anemias), renal failure (causing fluid retention), or overhydration. Interstitial edema is seen on chest radiographs and CT scans as blurring of the margins of the blood vessels and bronchial walls (peribronchial cuffing), thickening of the fissures (subpleural edema), and thickening of the interlobular septae (Kerley lines) (Fig. 3.11). As capillary pressure rises and interstitial pressure increases, water is forced into the alveolar spaces through the alveolar–capillary membrane; therefore, edema is often seen as a combination of both interstitial and alveolar opacities on the chest radiograph. The chest radiograph may also show associated findings of cardiomegaly, pleural effusions, widening of the vascular pedicle, enlargement of the azygos vein, and vascular redistribution (Fig. 3.12). Pulmonary edema is so common, relative to other causes of ILD, that it should often be considered the most likely diagnosis in the differential diagnosis of ILD. An uncommon pattern of edema is more common than an uncommon cause of ILD. Uncommon patterns of pulmonary edema can result from patient positioning or underlying perfusion abnormalities in the nonedematous lung (e.g., secondary to pulmonary embolism or asymmetric emphysema). Pulmonary edema can be caused by a number of processes other than chronic heart failure, and it may present with a normal-sized heart (Table 3.4).
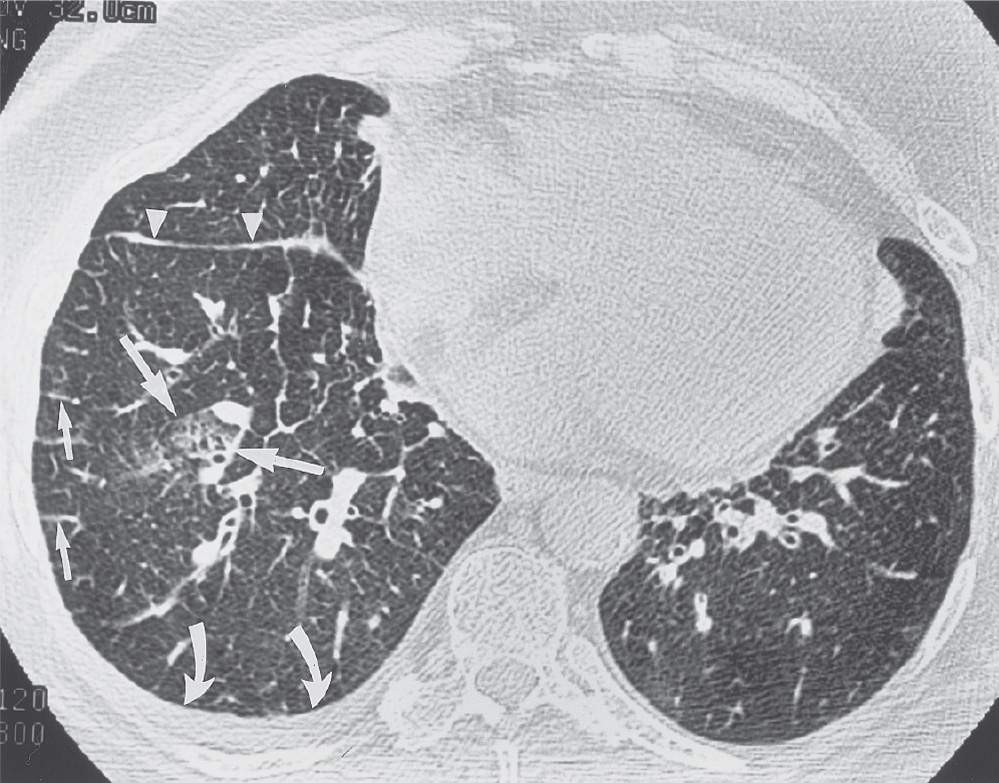
FIG. 3.11 • Cardiogenic pulmonary edema. This 69-year-old woman presented with left ventricular failure and a predominantly interstitial pattern of pulmonary edema. CT scan shows numerous Kerley B lines (short arrows), thickening of the right major fissure from subpleural edema (arrowheads), patchy areas of ground-glass opacification (long arrows), and a right pleural effusion (curved arrows).
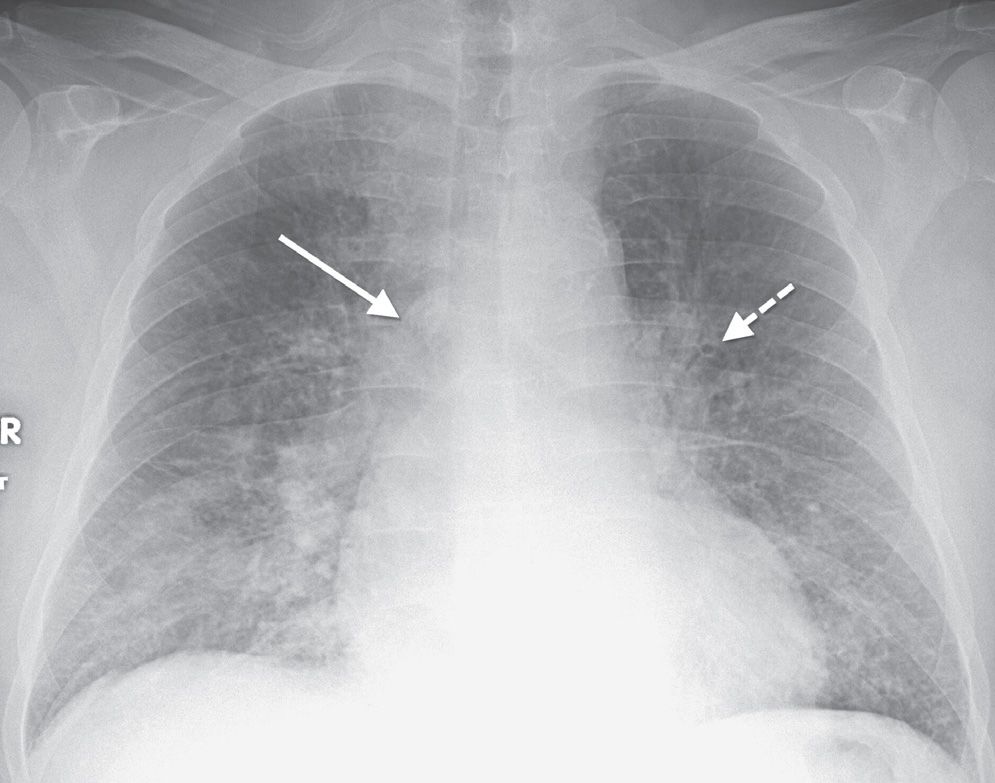
FIG. 3.12 • Cardiogenic pulmonary edema. PA chest radiograph shows enlargement of the cardiac silhouette, bilateral ILD, enlargement of the azygos vein (solid arrow), and peribronchial cuffing (dashed arrow).
Table 3.4 PULMONARY EDEMA WITH A NORMAL-SIZED HEART
“CHIHUAHUAH”
Central nervous system disorders
High-altitude pulmonary edema
Inhalation (e.g., carbon monoxide)
Heroin-induced
Uremia
Acute myocardial infarction
Hypersensitivity reaction
Underwater, near-drowning
Aspiration (gastric secretions)
Hemorrhage
IDIOPATHIC INTERSTITIAL PNEUMONIAS
The idiopathic interstitial pneumonias (IIPs) are a heterogeneous group of diffuse parenchymal lung diseases that have no well-defined cause (4). The classification is based on histologic criteria, although the diagnosis of IIP is made by correlating the clinical, imaging, and pathologic features. The morphologic, or “pattern,” of each IIP seen at histologic or CT examination is linked to a specific clinical syndrome. Clinical evaluation must prove that an interstitial pneumonia is idiopathic and exclude a recognizable cause (e.g., collagen vascular disease). Usual interstitial pneumonia (UIP) is the most common of the IIPs. Nonspecific interstitial pneumonia (NSIP) is the next most frequent. Cryptogenic organizing pneumonia (COP), desquamative interstitial pneumonia (DIP), respiratory bronchiolitis–associated interstitial lung disease (RB-ILD), and acute interstitial pneumonia (AIP) are less common, and lymphoid interstitial pneumonia (LIP) is rare. Typical CT features of each IIP are distinct, but there is overlap (Table 3.5). CT features of UIP and organizing pneumonia may be diagnostic in the correct clinical context, but those of NSIP, DIP, RB-ILD, AIP, and LIP are less specific.
UIP is characterized histologically by a patchy heterogeneous pattern with foci of normal lung, interstitial inflammation, fibroblastic proliferation, interstitial fibrosis, and honeycombing. Temporal heterogeneity is an important histologic feature and helps to distinguish UIP from DIP. Although the terms UIP and idiopathic pulmonary fibrosis (IPF) are often used interchangeably, the term IPF should be applied only to the clinical syndrome associated with the morphologic pattern of UIP. The typical CT features of UIP are a predominantly basal and subpleural reticular interstitial pattern with honeycombing and traction bronchiectasis (Fig. 3.13). Ground-glass opacity and consolidation can be seen but are not dominant features. Architectural distortion, reflecting lung fibrosis, is often prominent. In the correct clinical context, the CT features of UIP are often diagnostic. The presence of honeycombing as a predominant imaging finding is highly specific for UIP and can be used to differentiate it from NSIP, particularly when the distribution is patchy and subpleural-predominant (5). The presence of predominant ground-glass and reticular opacities is highly characteristic of NSIP, but there is a subset of patients with UIP who have this pattern and may require biopsy for differentiation from NSIP (Fig. 3.14). Distinction of UIP from other IIPs is important, because UIP is associated with a poorer prognosis than the other entities.
Table 3.5 IMAGING FEATURES OF IDIOPATHIC INTERSTITIAL PNEUMONIAS
Morphologic Pattern (Histologic and Radiologic) | Imaging Features |
UIP (clinical diagnosis of IPF) | Basal and subpleural–predominant distribution, reticular opacities (often with honeycombing), traction bronchiectasis, and architectural distortion |
NSIP (clinical diagnosis of NSIP) | Basal-predominant distribution, ground-glass and reticular opacities |
DIP (clinical diagnosis of DIP) | Basal and lower lung–predominant distribution, ground-glass opacities, sometimes with cysts |
Respiratory bronchiolitis (clinical diagnosis of RB-ILD) | Centrilobular distribution, ground-glass opacity, typically nodular |
Organizing pneumonia (clinical diagnosis of COP) | Basal and subpleural–predominant distribution, ground-glass opacity, and consolidation; bronchovascular distribution is also common |
Diffuse alveolar damage (clinical diagnosis of AIP) | Diffuse ground-glass opacity and consolidation |
LIP (clinical diagnosis of LIP) | Bronchovascular distribution is common, ground-glass and reticular opacities and perivascular cysts |
UIP, usual interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; DIP, desquamative interstitial pneumonia; RB-ILD, respiratory bronchiolitis–associated interstitial lung disease; COP, cryptogenic organizing pneumonia; AIP, acute interstitial pneumonia; LIP, lymphoid interstitial pneumonia.
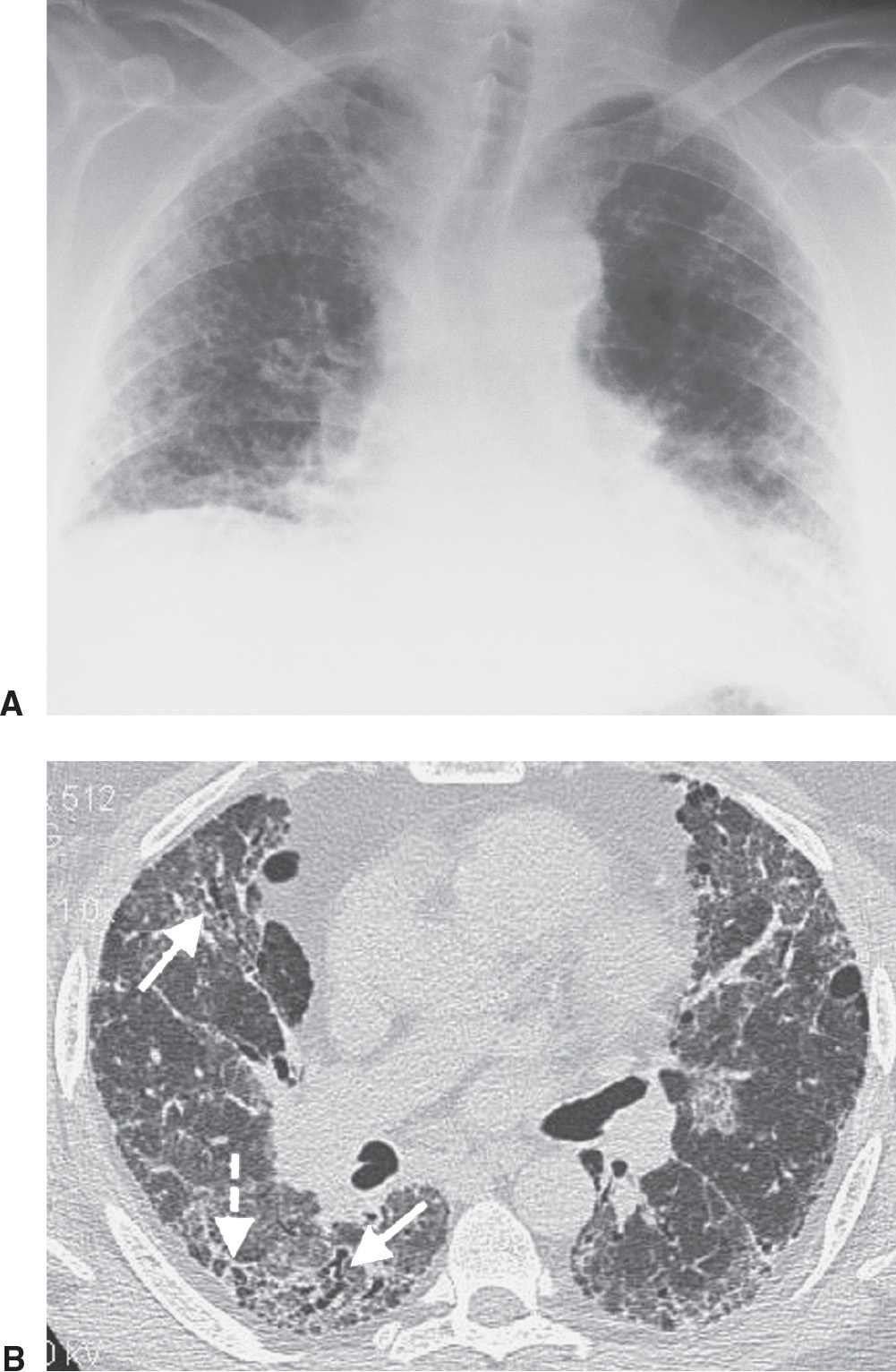
FIG. 3.13 • Usual interstitial pneumonia (UIP). A: PA chest radiograph shows medium to coarse reticular ILD with honeycombing, in a predominantly bibasilar and subpleural distribution. Lung volumes are decreased. B: CT scan shows bilateral subpleural honeycombing (dashed arrow), traction bronchiectasis (solid arrows), and a background of ground-glass opacity.
NSIP is characterized histologically by spatially homogeneous alveolar wall thickening caused by inflammation, fibrosis, or both. The spatial and temporal homogeneity of this pattern is important in distinguishing NSIP from UIP. The prognosis of NSIP is substantially better than that of UIP. Patients with NSIP are more commonly female and generally have a younger mean age than patients with UIP. The typical CT feature of NSIP is predominantly basilar ground-glass and reticular opacities (Fig. 3.15). Consolidation is uncommon and honeycombing is rare. The parenchymal abnormalities of NSIP may be reversible on follow-up CT scanning. Because the CT features of NSIP may overlap with those of organizing pneumonia, DIP, and UIP, a surgical lung biopsy should be considered when the CT pattern suggests NSIP (Fig. 3.16).
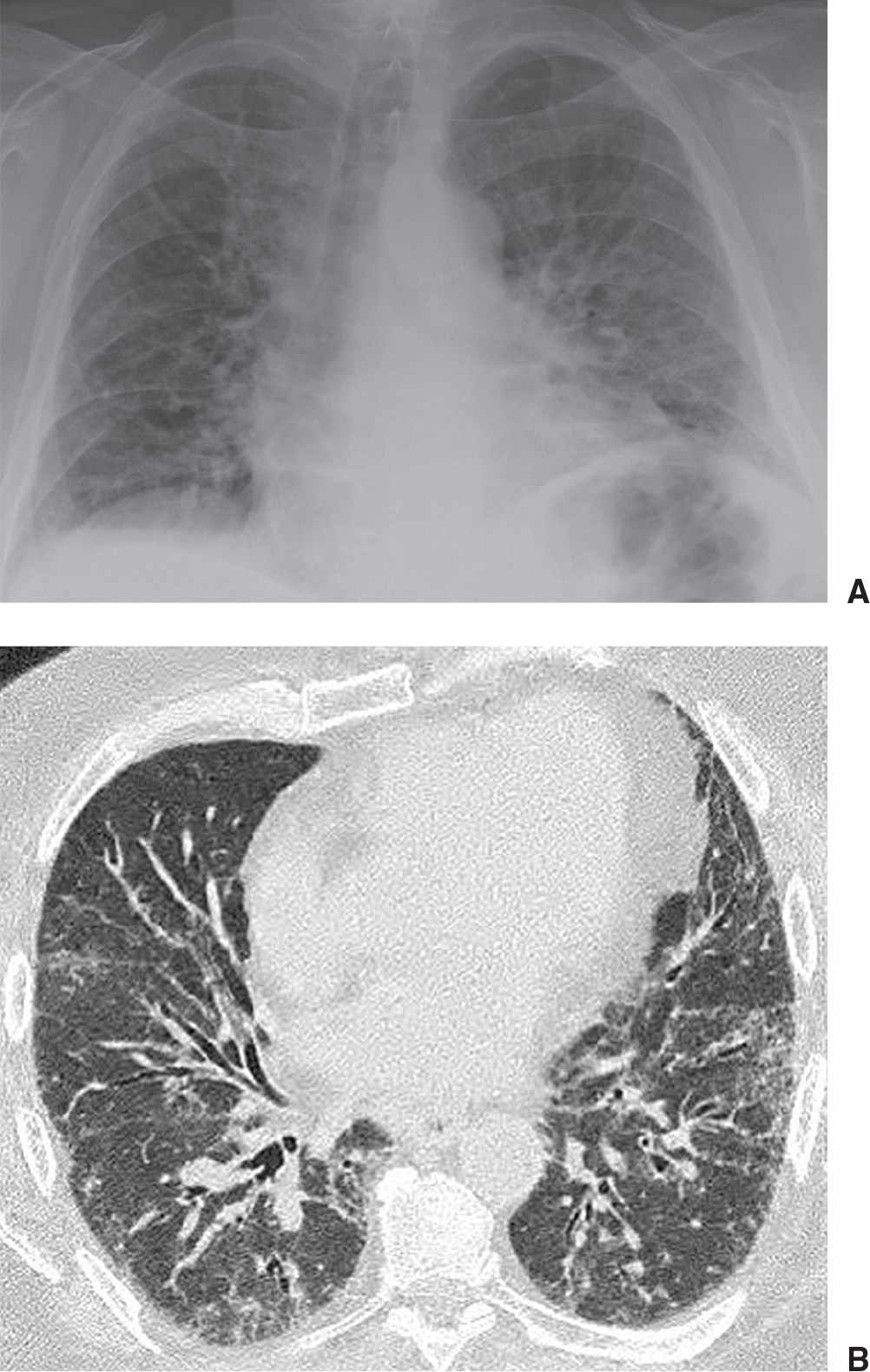
FIG. 3.14 • Systemic lupus erythematosus and UIP. A: PA chest radiograph shows low lung volumes and bibasilar reticular ILD. B: CT scan shows basilar subpleural ground-glass opacity and mild airway dilatation. Because a predominant pattern of honeycombing was not seen, lung biopsy was performed to confirm the diagnosis of UIP.
DIP is characterized histologically by spatially homogeneous thickening of alveolar septa, which is associated with intra-alveolar accumulation of macrophages. The term desquamative refers to an initially incorrect belief that the intra-alveolar macrophages represented desquamated alveolar cells. The majority of patients are cigarette smokers in their fourth or fifth decade of life (6). DIP is more common in men than in women. Most patients improve with cessation of smoking and oral corticosteroids. The histologic features of DIP are similar to those of RB-ILD (a condition seen exclusively in smokers), although the distribution of DIP is diffuse and RB-ILD has a predominantly bronchiolocentric distribution. The typical CT feature of DIP is ground-glass opacity in a predominantly lower lung distribution (Figs. 3.17 and 3.18). Reticulation is frequently seen but is typically limited to the lung bases. Well-defined cysts can occur within the areas of ground-glass opacity.
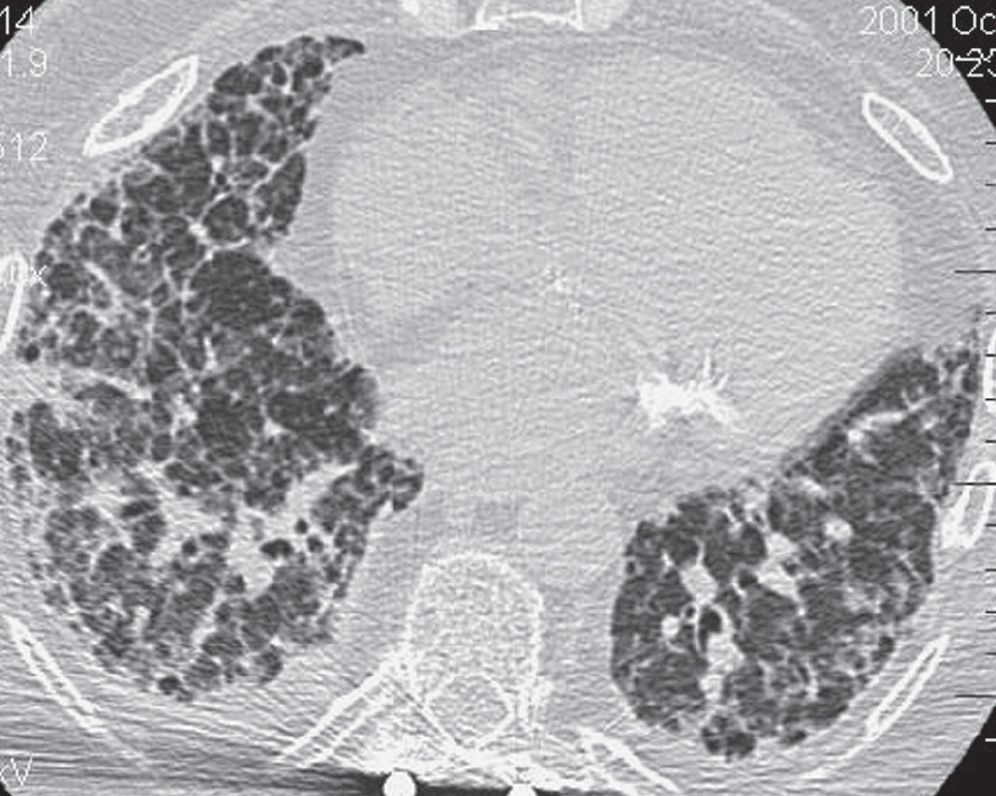
FIG. 3.15 • Nonspecific interstitial pneumonia (NSIP). CT scan shows bibasilar reticular and ground-glass opacities.
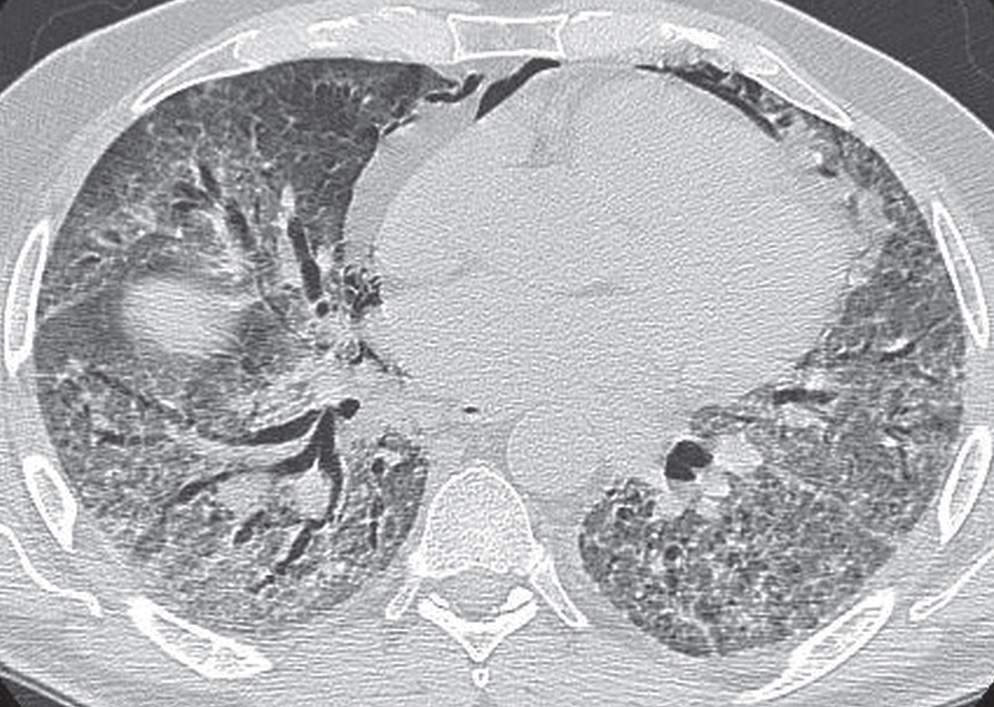
FIG. 3.16 • Nonspecific interstitial pneumonia. CT scan of a 61-year-old man with increasing cough, shortness of breath, and chest pain shows bibasilar reticular and ground-glass opacities.
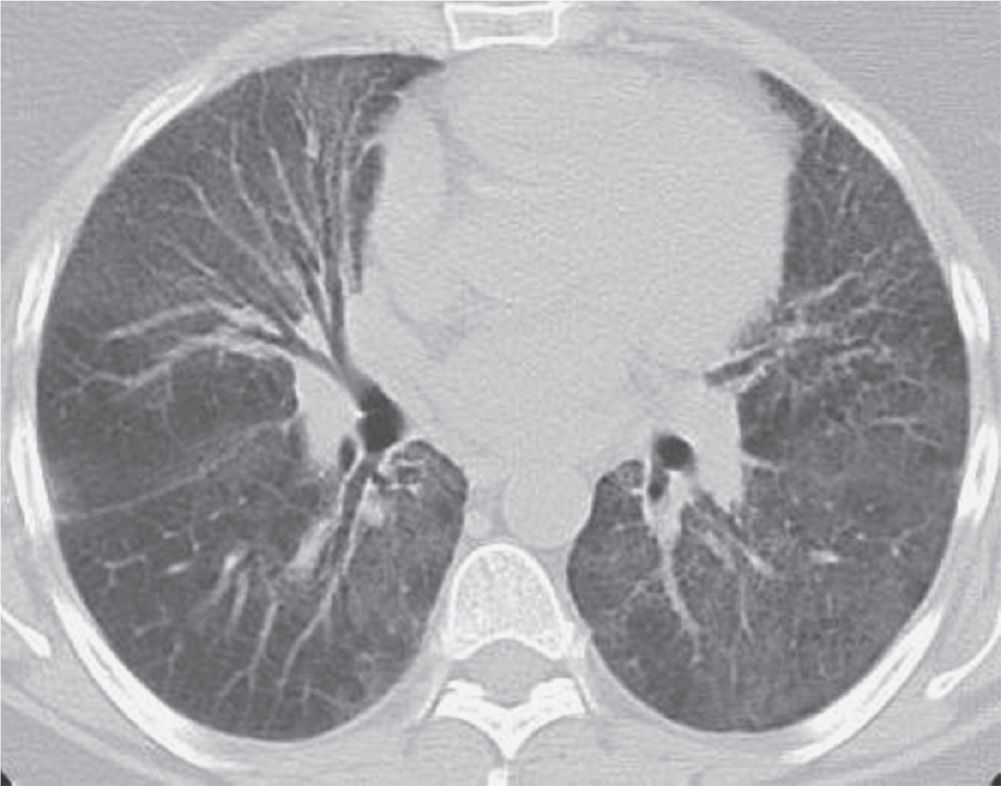
FIG. 3.17 • Desquamative interstitial pneumonia (DIP). CT scan shows bilateral ground-glass opacity in a predominantly lower lung distribution.
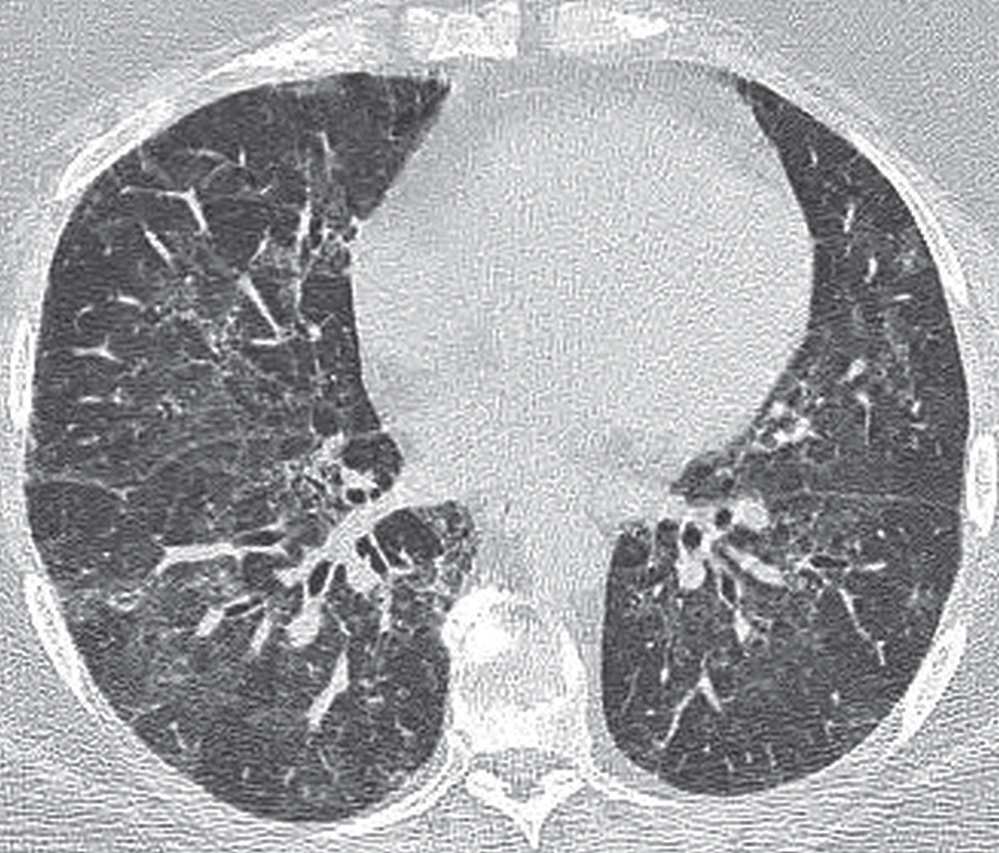
FIG. 3.18 • Desquamative interstitial pneumonia (DIP). CT scan shows bilateral ground-glass opacity in a predominantly lower lung distribution.
Respiratory bronchiolitis is a histopathologic lesion found in cigarette smokers and is characterized by the presence of pigmented intraluminal macrophages within respiratory bronchioles (4). It is usually asymptomatic. In rare cases, patients who are heavy smokers may develop RB-ILD, a condition characterized by pulmonary symptoms, abnormal pulmonary function, and imaging abnormalities, with respiratory bronchiolitis being the only histologic lesion identified on lung biopsy. Respiratory bronchiolitis, RB-ILD, and DIP are regarded as a continuum of smoking-related lung injuries. The CT features of patients with asymptomatic respiratory bronchiolitis show ground-glass centrilobular nodules and patchy areas of ground-glass opacity (Fig. 3.19). In RB-ILD, the findings are more extensive (Fig. 3.20) but are at least partially reversible in patients who stop smoking. The imaging features of RB-ILD may be similar to those of hypersensitivity pneumonitis and NSIP. Patients with hypersensitivity pneumonitis often have a history of exposure to an inciting agent and are usually nonsmokers.
Although COP is primarily an intra-alveolar process, it is included in the classification of the IIPs because of its idiopathic nature and because its appearance may overlap with that of the other IIPs. The term organizing pneumonia refers to the morphologic imaging or histologic pattern (associated with a wide variety of diseases), whereas COP indicates the associated idiopathic clinical syndrome. Histologically, organizing pneumonia is distinguished by patchy areas of consolidation characterized by polypoid plugs of loose organizing connective tissue with or without endobronchiolar intraluminal polyps. The architecture of the lung is preserved. Patients with COP typically present with cough and dyspnea of relatively short duration. Consolidation is present on CT images in 90% of patients with COP, with a subpleural or peribronchial distribution in up to 50% of cases (4) (Figs. 3.21 and 3.22). Air bronchograms, with mild cylindric bronchial dilatation, are common. Ground-glass opacities are present in about 60% of cases. The lower lungs are more frequently involved. Findings usually improve with steroid treatment. The differential diagnosis of COP includes adenocarcinoma in situ (formerly BAC), lymphoma, vasculitis, sarcoidosis, chronic eosinophilic pneumonia, and infectious pneumonia.
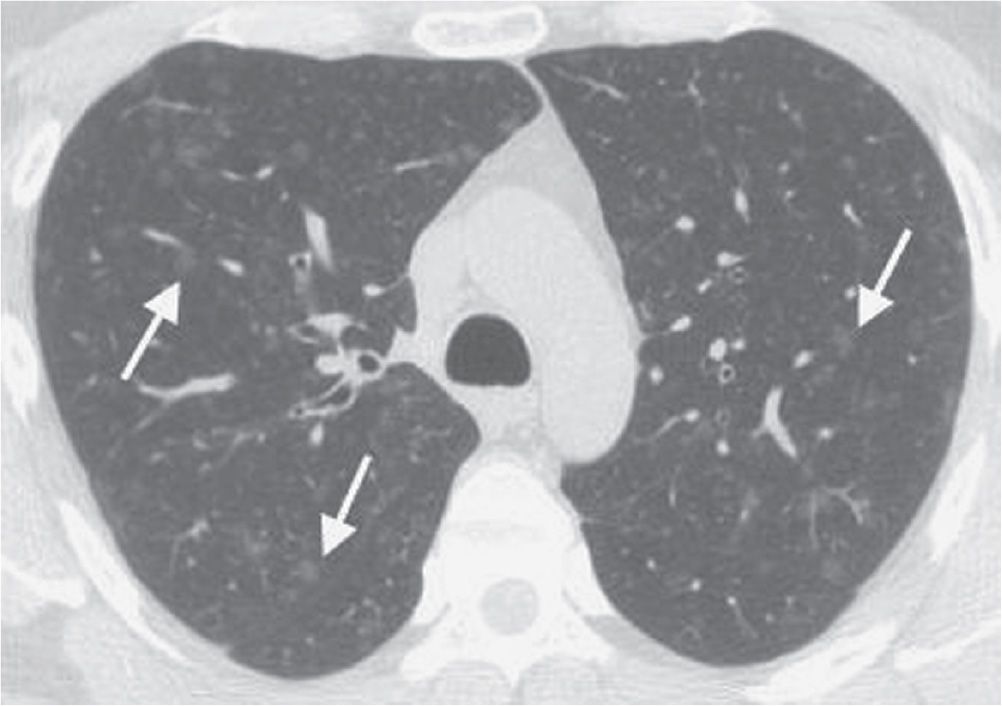
FIG. 3.19 • Respiratory bronchiolitis. This patient had a long history of cigarette smoking and no respiratory symptoms. CT scan shows numerous ground-glass nodules in a centrilobular distribution (arrows).
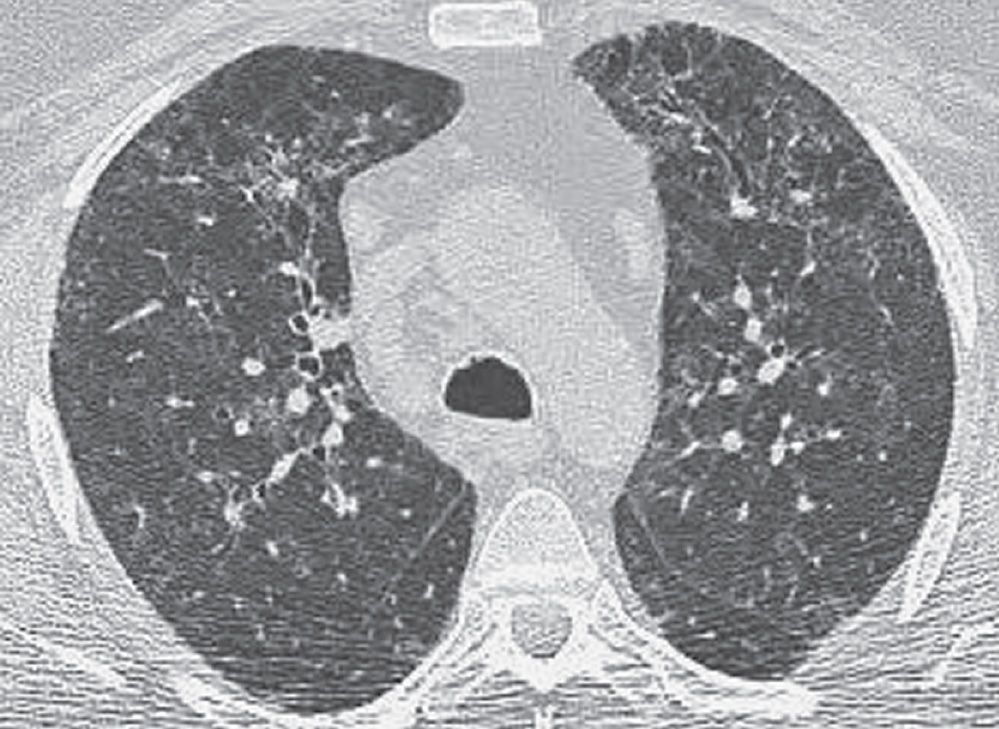
FIG. 3.20 • Respiratory bronchiolitis-associated interstitial lung disease (RB-ILD). This patient had a long history of cigarette smoking, chronic cough, and shortness of breath. CT scan shows bilateral reticular and ground-glass opacities in a predominantly upper lung distribution.
AIP is a rapidly progressive form of interstitial pneumonia characterized histologically by hyaline membranes within the alveoli and diffuse, active interstitial fibrosis indistinguishable from the histologic pattern found in acute respiratory distress syndrome caused by sepsis and shock. The term AIP is reserved for diffuse alveolar damage of unknown origin. Patients with AIP present with respiratory failure developing over days or weeks. Mechanical ventilation is usually required. No etiologic agent is identified. Typical CT features of early-stage AIP are ground-glass opacity, bronchiolar dilatation, and dense airspace opacity. Late-stage features are honeycombing, architectural distortion, and traction bronchiectasis.
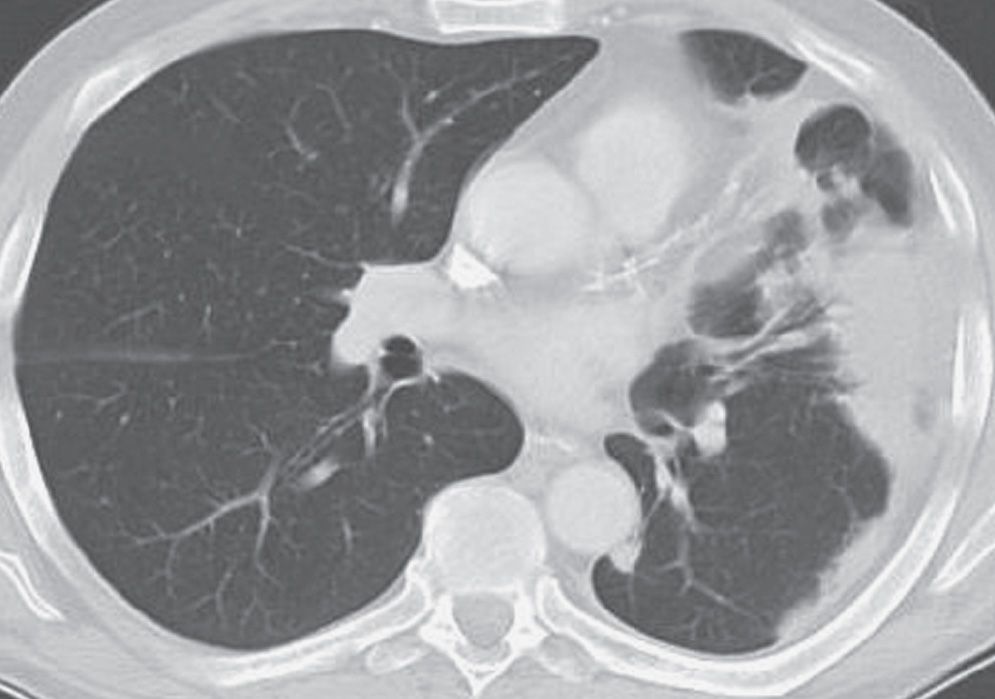
FIG. 3.21 • Organizing pneumonia. This patient presented with acute shortness of breath and nonproductive cough. CT scan shows subpleural, dense airspace opacity in the left lung.
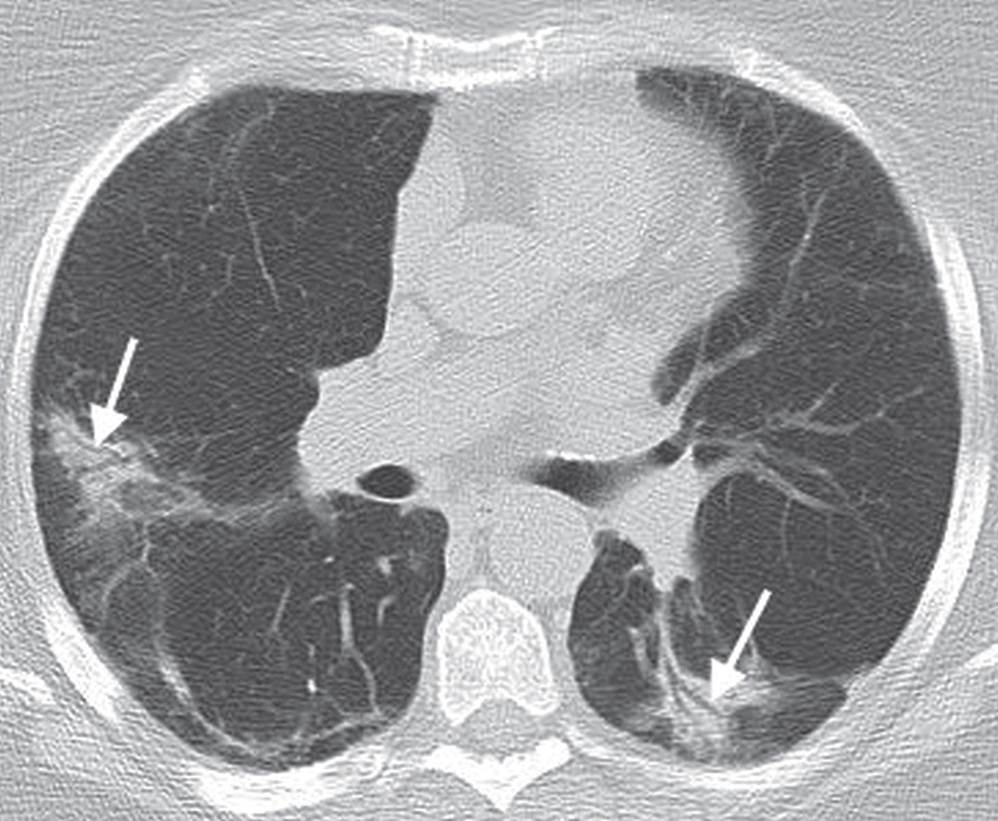
FIG. 3.22 • Organizing pneumonia. CT scan of a 74-year-old man with cough shows patchy ground-glass opacity in a bronchovascular distribution (arrows).
In adults, LIP is commonly associated with connective tissue disorders (particularly Sjögren syndrome), immunodeficiency syndromes, and Castleman syndrome. Idiopathic LIP is rare. The histologic feature of LIP is alveolar septal interstitial infiltration by lymphocytes and plasma cells. The typical CT findings are ground-glass and reticular opacities, sometimes associated with perivascular cysts (Fig. 3.23). Other findings may include lung nodules, dense airspace opacity, thickening of the bronchovascular bundles, and interlobular septal thickening.
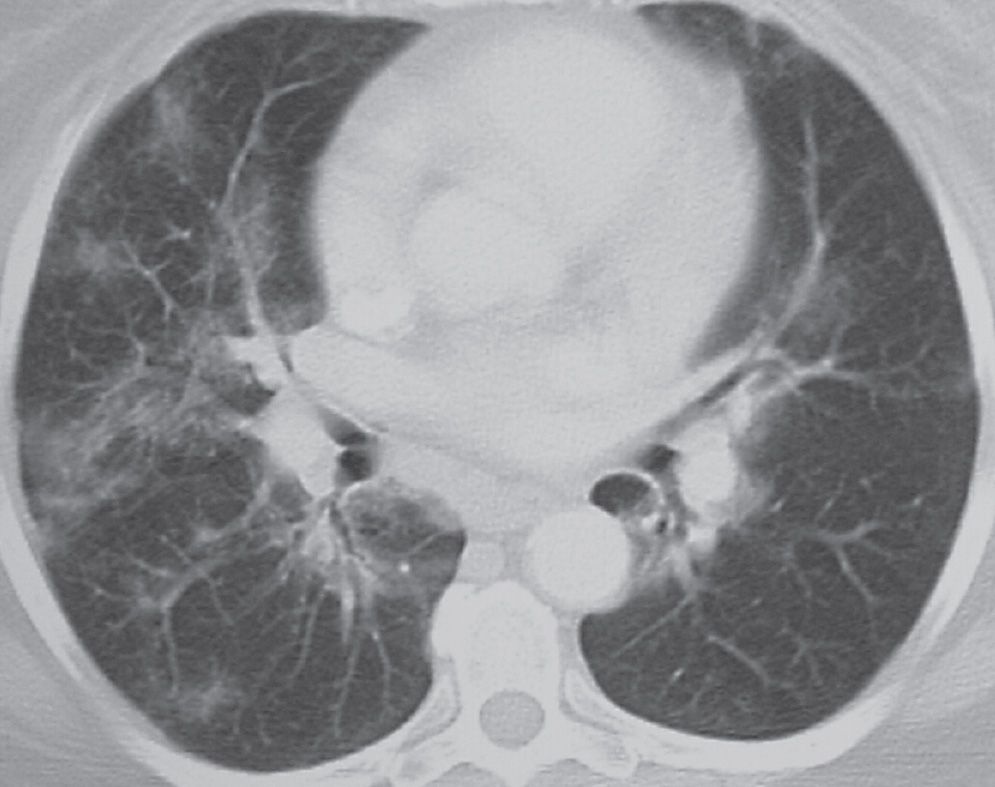
FIG. 3.23 • Lymphoid interstitial pneumonia (LIP). This patient had Sjögren syndrome and new respiratory symptoms. CT scan shows bilateral patchy ground-glass opacities in a bronchovascular distribution.
INFECTIOUS INTERSTITIAL PNEUMONIA
Infectious pneumonia resulting in a diffuse interstitial pattern is unusual; however, viral, fungal, mycobacterial, and Mycoplasma pneumonias may be predominantly interstitial or interstitial-appearing. Fungal disease is discussed in Chapter 7. Pneumocystis pneumonia also produces a fine interstitial pattern on chest radiography, and is discussed in Chapter 16.
Mycoplasma pneumoniae usually affects previously healthy individuals between the ages of 5 and 40 years (7). Chest radiographs may show widespread bilateral nodular or reticular opacities, and they may take several weeks to return to normal. Alternatively, dense airspace opacity may be seen involving one or several lobes.
Viruses are the major cause of respiratory tract infection in the community, especially in children. The most common viral pneumonias in infants and young children are caused by respiratory syncytial virus, parainfluenza virus, adenovirus, and influenza; in adults, influenza and adenovirus are most common. Viruses that cause pneumonia in immunocompromised patients include Cytomegalovirus, varicella-zoster, and herpesvirus. The radiographic appearance of viral pneumonias is typically a diffuse interstitial pattern with a diffuse, patchy, often nodular appearance (Fig. 3.24).
DRUG TOXICITY
Numerous drugs, some of which are listed in Table 3.6, can result in transient or permanent lung injury of varying types and severities (Fig. 3.25). A more complete list can be found in the medical literature (8). Diffuse alveolar damage is a common manifestation of pulmonary drug toxicity and is frequently caused by cytotoxic drugs, especially cyclophosphamide, bleomycin, and carmustine. It manifests radiographically as bilateral hetero- or homogeneous opacities usually in the mid- and lower lungs and on CT scans as scattered or diffuse areas of ground-glass opacity. NSIP occurs most commonly as a manifestation of carmustine toxicity or of toxicity from noncytotoxic drugs such as amiodarone. At radiography, it appears as diffuse areas of heterogeneous opacity, whereas early CT scans show diffuse ground-glass opacity, and late CT scans show fibrosis in a basal distribution. Organizing pneumonia, which is commonly caused by bleomycin and cyclophosphamide (as well as gold salts and methotrexate), appears on radiographs as hetero- and homogeneous peripheral opacities in both upper and lower lobes and on CT scans as poorly defined nodular consolidation, centrilobular nodules, and bronchial dilatation. Other manifestations of pulmonary drug toxicity include eosinophilic pneumonia, constrictive bronchiolitis, pulmonary hemorrhage, edema, hypertension, and veno-occlusive disease.
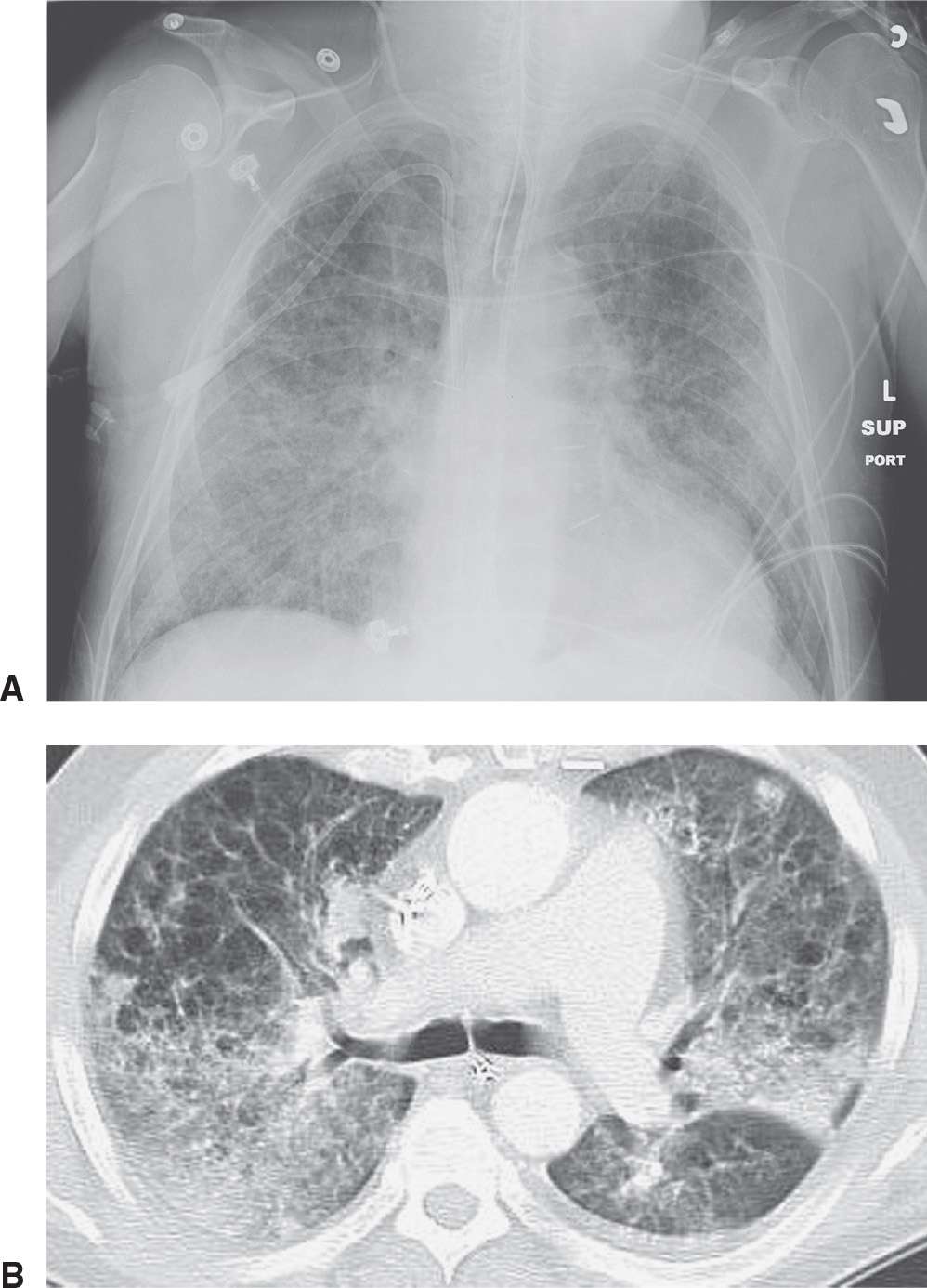
FIG. 3.24 • Influenza pneumonia. This patient had a history of emphysema and acute respiratory symptoms. A: Supine chest radiograph shows bilateral reticular ILD. B: CT scan shows bilateral reticular and ground-glass opacities and areas of consolidation. “Cystic” areas represent pulmonary emphysema.
Table 3.6 COMMONLY USED DRUGS THAT CAN CAUSE LUNG TOXICITY
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


