5 Intracranial Tumors
Among the many intra-axial and extra-axial brain tumors that are known, only a few types account for the majority of forms that are observed clinically. It should also be considered that the differential diagnosis of brain tumors in emergency situations is of less importance than detecting a potentially devastating mass effect involving a shift of the midline or a sub-falcial/transtentorial herniation. Magnetic resonance imaging (MRI) is generally superior to computed tomography (CT) for tumor differentiation and is the modality of choice for that application. The approximate prevalences of the various tumor types are reviewed in Table 5.1.
| Tumor | Prevalence (%) |
|---|---|
| Gliomas | 50 |
| Meningioma | 10 |
| Pituitary adenoma | 7 |
| Neuroma | 7 |
| Craniopharyngioma, dermoid, epidermoid, teratoma | 4 |
| Angioma | 4 |
| Sarcoma | 4 |
| Other | 14 |
| Total | 100 |
* Cited after Adams RD, Victor M, Principles of Neurology (New York: McGraw-Hill, 3rd ed. 1985), based on a series of approximately 15000 patients reviewed by Zülch, Cushing, and Olivecrona. The authors report the following figures for children: astrocytoma 48%, medulloblastoma 44%, ependymoma 8%.
Neuroepithelial Tumors
Frequency: gliomas account for approximately half of all intracranial tumors. Glioblastoma (grade IV glioma) predominates, accounting for about half of all gliomas. Low-grade astrocytomas account for 30%, and all others (oligodendroglioma, pilocytic astrocytoma) make up less than 10%.
Suggestive morphologic findings: white-matter hypodensity (low-grade forms) or mass with scalloped rim enhancement (higher-grade forms).
Procedure: biopsy confirmation and surgical treatment.
Other studies: MRI defines the tumor extent more accurately than CT. Positron-emission tomography (PET) is superior for detecting recurrence. CT is the method of choice for oligodendroglioma, due to frequent calcification.
Checklist for scan interpretation:
 Extent, edema, mass effect?
Extent, edema, mass effect?
 Spread to contralateral hemisphere?
Spread to contralateral hemisphere?
 Calcifications?
Calcifications?

The “glial subgroup of neuroepithelial tumors,” as gliomas are called in the World Health Organization (WHO) classification, consists of the following types:
Astrocytoma. Astrocytomas are gliomas derived from astrocytes. The subtypes in the WHO classification are indistinguishable by their imaging features.
Anaplastic astrocytoma. Anaplastic astrocytomas are grade III neoplasms characterized by rapid progression, transformation to glioblastoma, and enhancement on postcontrast CT.
Glioblastoma. Glioblastomas display a heterogeneous CT pattern of necrosis, hemorrhage, and contrast enhancement. They are classified according to their biologic behavior as grade IV gliomas. The histologic subtypes are indistinguishable by CT.
Pilocytic astrocytoma. Pilocytic astrocytomas are characterized by less infiltrative growth, a low propensity for malignant transformation, a protracted course, and a peak age incidence in adolescents. They are classified as grade I gliomas, which very rarely undergo malignant transformation to anaplastic pilocytic astrocytoma (grade III). CT can provide a presumptive diagnosis.
Pleomorphic xanthoastrocytoma. This tumor is a variant commonly found in patients with seizure disorder. With its relative lack of aggressiveness, it is classified as a grade II glioma. Malignant transformation is rare but can occur.
Subependymal giant-cell astrocytoma. This tumor occurs in tuberous sclerosis and has a characteristic CT appearance. Histologically, it is a grade I glioma.
Oligodendroglioma. Calcifications characterize both the histologic and CT appearance of oligodendrogliomas. These tumors often arise in the white matter or basal ganglia, and have a strong tendency to infiltrate the cortex and leptomeninges. Oligodendrogliomas are grade II neoplasms that have less malignant potential than astrocytomas.
Anaplastic oligodendroglioma. Anaplastic oligodendrogliomas are grade III gliomas. Malignant transformation to grade IV is usually indistinguishable from glioblastoma.
Grading of gliomas. The typical CT appearance of gliomas varies with the histologic grade of the neoplasm:
• Grade I gliomas. Grade I gliomas are characterized by their location (cerebellum, pons, optic tract), age of occurrence (mainly children and adolescents), and the CT appearance of a cyst and/or an enhancing tumor nodule (Figs. 5.1, 5.2). Subependymal giant-cell astrocytomas belong to this category.
• Grade II gliomas. These tumors usually appear as a hypodense area that exhibits little or no mass effect and no contrast enhancement. The absence of calcifications suggests astrocytoma, while the presence of calcifications is more consistent with oligodendroglioma (Figs. 5.3–5.5). Special forms include pleomorphic xanthoastrocytoma and gemistocytic astrocytoma.
• Grade III gliomas. Contrast enhancement occurs in portions of grade III gliomas (Figs. 5.6–5.8). Anaplastic astrocytomas and anaplastic oligodendrogliomas are observed.
• Grade IV gliomas. These tumors are characterized by a heterogeneous CT pattern with necrosis, hemorrhage, contrast enhancement, perifocal edema, and a scalloped or irregular enhancing rim. The histologic subtypes are glioblastoma, anaplastic astrocytoma, and anaplastic oligodendroglioma (Figs. 5.9, 5.10, 5.15).
In addition to these forms, there are also mixed gliomas in which two or more neoplastic glial cell types coexist in the same tumor, such as oligoastrocytoma and anaplastic oligoastrocytoma.
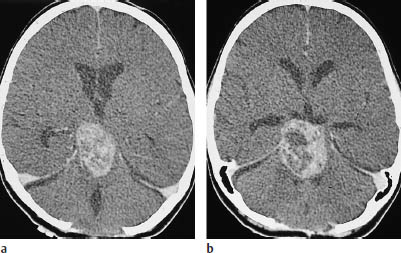
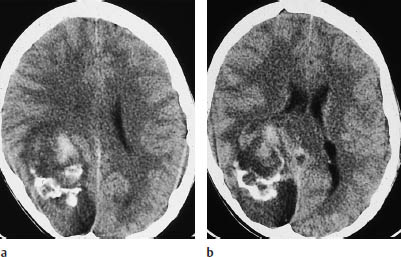
Fig. 5.2a, b Grade I glioma. The grade I gliomas found in children consistently enhance after contrast administration, but may also show extensive necrosis. In the case of this 14-year-old boy, contrast enhancement occurs in the rostral portions of the tumor. Calcifications are also visible within the hypodense glioma. There is a considerable mass effect, with anterior displacement of the right occipital horn.
 Frequency
Frequency
The incidence of supratentorial brain tumors correlates with their mortality rates, and considerable variation is seen in the geographic and histologic distribution of occurrence. For example, age-corrected incidence rates range from 1.5 per 100,000 (Singapore) to as much as 9.1–10 per 100,000 (Sweden and Israel). Incidence rates are generally higher in countries with a high socioeconomic status. A comparison of mortality rates in different countries shows a very broad range, from 1.1 per 100,000 in Mexico (1951–1958) to 10 per 100,000 in the Federal Republic of Germany (1967–1973). Rising mortality rates have been confirmed in a number of epidemiologic studies. The incidence of brain tumors in the United States is estimated at approximately 12,000 new cases per year.
The following statistical distribution has been calculated for the different grades of glioma:
Glioblastoma: anaplastic astrocytoma: low-grade astrocytoma = 5 : 3 : 2.
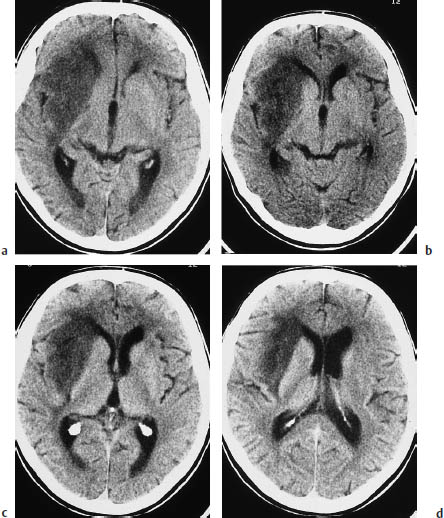
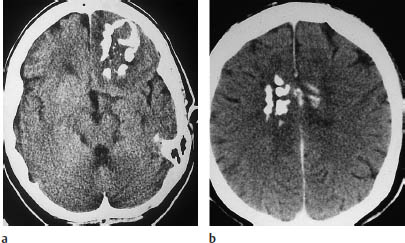
Fig. 5.4 Oligodendroglioma. Most oligodendrogliomas can be identified on CT by their calcifications. The two scans show typical coarse, irregular calcifications in the left frontal lobe and in the rostral portion of the corpus callosum. White-matter hypodensity is a far less important morphologic feature of oligodendroglioma than calcifications.
a Calcifications in the left frontal lobe.
b Calcifications in the corpus callosum.
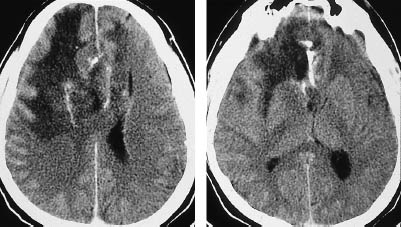
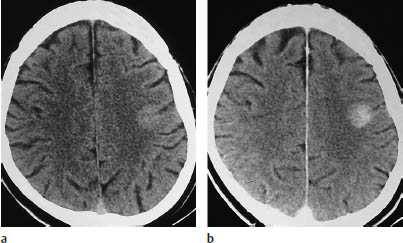
Fig.5.6 Anaplastic astrocytoma. Anaplastic astrocytomas are classified as grade III gliomas and show more or less intense contrast enhancement. Noncontrast CT shows a small lesion in the left parietal area, the attenuation of which is closer to that of the cerebral cortex than the white matter. Moderate enhancement occurs after contrast administration.
a Noncontrast scan.
b Postcontrast scan.
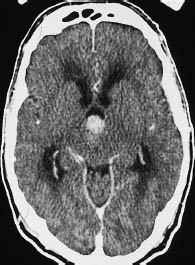
Fig. 5.7 Grade III anaplastic astrocytoma. CT shows a primary intraventricular brain tumor. This type of tumor can lead to ventricular expansion and pressure coning.
Fig. 5.8 Grade III glioma. Grade III gliomas are indistinguishable from glioblastoma multiforme by their CT appearance. A tumor showing intense, scalloped rim enhancement is visible lateral to the left occipital horn.
a Noncontrast scan.
b Postcontrast scan.
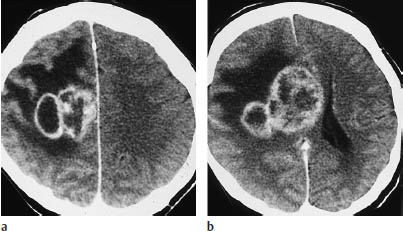
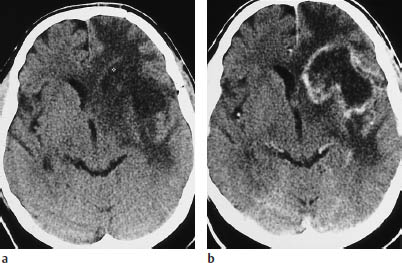
Fig. 5.10a, b Glioblastoma. A scalloped or multi-ring pattern of contrast enhancement is characteristic of glioblastoma. These scans show extensive edema, involving almost all of the white matter in the right hemisphere.
 Clinical Manifestations
Clinical Manifestations
Pilocytic astrocytomas occur predominantly in children and young adults. The peak age of occurrence is between 40 and 50 for astrocytomas and oligodendrogliomas, and somewhat later for glioblastomas. All higher-grade gliomas (III or IV) tend to occur at age 20 or older, and pilocytic astrocytomas may be diagnosed after age 20.
 CT Morphology
CT Morphology
Pilocytic astrocytoma. This tumor may occur in the cerebellum, brain stem, and less commonly at supratentorial sites. Infratentorial tumors can be distinguished from midline medulloblastoma by their eccentric location. CT without intravenous contrast shows an ill-defined hypodense lesion with a mass effect or a corresponding cyst. Postcontrast CT often demonstrates nodular enhancement in the cyst wall.
Optic glioma. Most grade II-IV gliomas are supratentorial and appear as hypodense masses in the white matter. Only oligodendroglioma shows a predilection for the frontal or temporal lobe and infiltrates the cerebral cortex. The margins are ill-defined. Mass effects range from the effacement of individual sulci and subtle frontal-horn displacement to a shift of the midline and ventricular compression. The calcifications found in oligodendrogliomas are more common in lower-grade tumors than in the more malignant forms. The calcifications may be flocculent or stippled, and may follow the outline of the cerebral cortex. Contrast enhancement occurs in grade III and grade IV gliomas. The enhancement in grade III gliomas shows a patchy or bizarre pattern, while grade IV gliomas show a scalloped or irregular enhancing rim with central necrosis. Grade IV gliomas typically show a heterogeneous mix of enhancing and hypodense tumor tissue, necrosis or cysts, intratumoral hemorrhage, and mass effect.
Butterfly glioma. Butterfly glioma is a bihemispheric tumor that arises predominantly in the anterior or posterior commissure and infiltrates both hemispheres (Fig. 5.11).
Gliomatosis cerebri, or diffuse gliomatosis. Gliomatosis cerebri refers to a generalized malignant transformation that may involve all of the cerebral white matter. The course may span many years, and a mass effect may be absent or limited to mild compression of the frontal or occipital horns.
Multifocal gliomas. This is a pattern characterized by synchronous or metachronous development of multiple tumor foci (Fig. 5.12).
 Differential Diagnosis
Differential Diagnosis
The imaging appearance of cerebral infarction most closely resembles that of gliomas (Fig. 5.13).
With cerebral infarction, however, the distribution of the hypodense area often conforms to a vascular territory or to a watershed area between two territories. Also, infarctions tend to involve the adjacent cortical gray matter more extensively, and they show predominantly cortical enhancement after contrast administration. Infarcts have an abrupt clinical onset that contrasts with the typically insidious onset of gliomas. Serial examinations after an infarction usually show a regression of changes with fading enhancement, whereas gliomas show either no significant change (low-grade forms) or marked progression (high-grade forms). As for inflammatory lesions, multiple sclerosis (Fig. 5.14) and granulomas (e.g., in tuberculosis) can sometimes mimic the neuroimaging features of higher-grade gliomas.
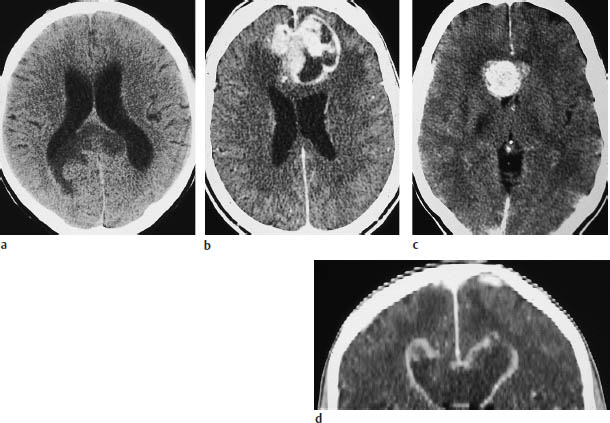
Fig. 5.11 a–d Butterfly glioma. “Butterfly glioma” is the term applied to primary brain tumors that extend across the anterior or posterior commissure or corpus callosum to involve both hemispheres.
a The noncontrast scan shows a low-grade glioma of the posterior commissure.
b This postcontrast scan in a different patient shows a glioblastoma with a scalloped, enhancing rim.
c Corpus callosum glioma may also appear as a solid round mass.
d The coronal reformatted image shows two ring-like masses located above the lateral ventricles.
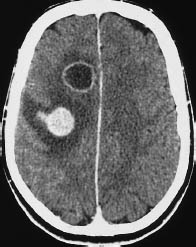
Fig. 5.12 Glioblastoma. Individual glioblastomas can exhibit multiple tumor foci. This scan illustrates one ring-enhancing lesion and one nodular-enhancing lesion with perifocal edema. Similar patterns may be seen with metastases. However, it should be carefully checked whether there are bridging parts of the tumor connecting between lesions that appear to be solitary.
In the case of multiple sclerosis, examination of the cerebrospinal fluid (CSF) will help to narrow the differential diagnosis. It is rare, however, for multiple sclerosis to present with a large enhancing lesion, extensive perifocal edema, and an acute onset. In the case of metastases, multiplicity may not be a reliable differentiating criterion due to the potential multicentric occurrence of glioblastoma.
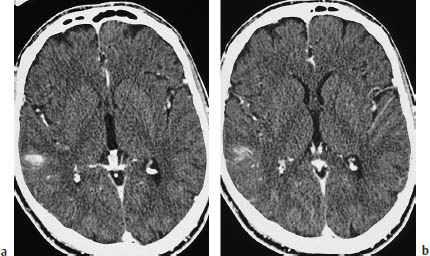
Fig.5.13a, b Primary brain tumor mimicking cerebral infarction. If the diagnosis is made promptly, there should be little danger of confusing primary brain tumors with infarction. These scans show a primary brain tumor that is manifested only by mild, ill-defined contrast enhancement in the right parietal
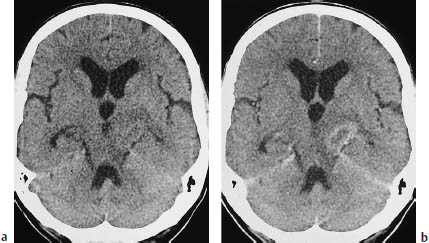
Fig. 5.14a, b Primary brain tumor mimicking a metastasis or demyelinating lesion. The CT morphology of primary brain tumors can also mimic that of metastases. This tumor, located in the left cerebral peduncle, appears hypodense on plain CT and shows faint ring enhancement after contrast administration.
a Noncontrast scan.
b Postcontrast scan.
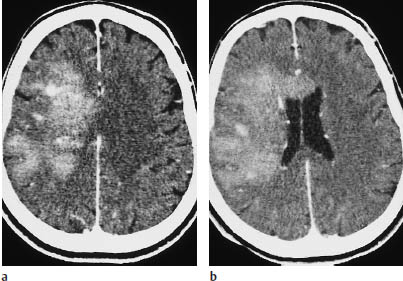
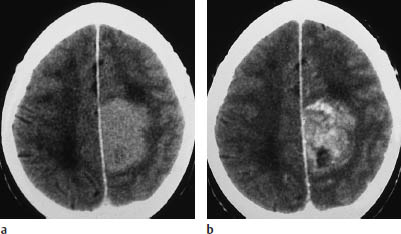
Fig. 5.16a, b Parasagittal astrocytoma. This parasagittal astrocytoma shows intense enhancement after contrast administration. It could be mistaken for a meningioma.
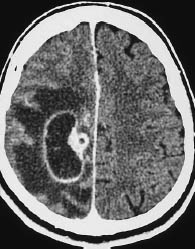
Fig. 5.17 Glioblastoma. The scalloped enhancing rim of glioblastoma can mimic other tumors in certain CT slices. Location aside, the enhancing rim, central necrosis, and enhancing nodular component of this glioblastoma could also suggest angioblastoma, although it is extremely rare for that type of tumor to occur at the supratentorial level.
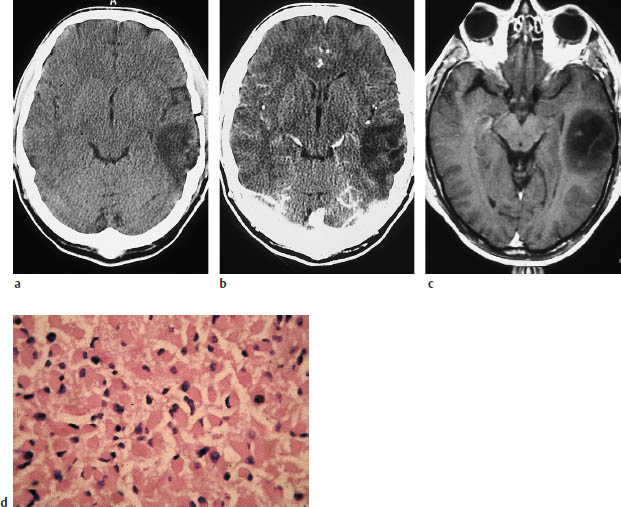
a, b Noncontrast CT (a) and postcontrast CT (b) show a relatively well-marginated, hypodense tumor in the left temporal lobe that does not enhance after contrast administration.
c The postcontrast magnetic resonance image also shows no enhancement.
d Typical histologic pattern, with plump eosinophilic cells and eccentric pleomorphic nuclei.
Even when all morphologic criteria have been evaluated, there will still be a certain percentage of cases in which only stereotactic biopsy can establish the diagnosis. The diverse imaging findings can lead to numerous misinterpretations, but these can be minimized by keeping in mind the frequency of glioblastoma.
 Follow-Up
Follow-Up
Two factors should be considered in the radiologic follow-up of brain tumor patients who have undergone surgical resection or radiotherapy:
• Both surgery and radiotherapy induce changes that can mimic a residual or recurrent tumor.
• Steroid therapy, by stabilizing defects in the blood-brain barrier, tends to reduce the size of perifocal edema and of enhancing tumor elements.
When a tumor has been surgically removed, the granulating resection margins will usually start to enhance on about the fifth postoperative day. This enhancement may persist for months. Thus, only an immediate postoperative examination can provide reliable information regarding residual tumor (assuming that the tumor enhanced prior to surgery).
Modern techniques of aggressive, selective radiotherapy can be very successful in inducing radiation necrosis at the tumor site. This necrotic area enhances after intravenous contrast administration and should not be mistaken for residual or recurrent tumor. The enhancement usually appears at about 4–8 months after the end of treatment and typically shows a ring pattern that is slightly larger than the original tumor.
It should be noted that perifocal edema and contrast enhancement may be decreased in volume and prominence by the initiation of steroid therapy, or by administering steroids at an excessive dosage. Consequently, these agents should be administered at a constant dosage for some time in order to allow accurate assessment of tumor regression or progression.
As for CT findings in patients who do not develop a recurrence, leukoencephalopathic changes and signs of cerebral atrophy begin to appear 6 months after surgery or radiotherapy. The CT hallmark of leukoencephalopathy is an accentuation of gray-white matter contrast due to a generalized decrease in white-matter density.
Pleomorphic Xanthoastrocytoma
Frequency: very rare.
Suggestive morphologic findings: hypodense mass located in the cortex or at the gray-white matter junction, often containing both cystic and solid components.
Procedure: treatment consists of surgical resection.
Other studies: as with all primary brain tumors, MRI is more sensitive than CT.
Checklist for scan interpretation:
 Location (precentral/postcentral)?
Location (precentral/postcentral)?
 Evidence of dural involvement?
Evidence of dural involvement?
 Pathogenesis
Pathogenesis
Pleomorphic xanthoastrocytoma (Fig. 5.19) is a special form of astrocytoma that involves the cortex and leptomeninges. The prognosis is relatively good. Some cases of subarachnoid dissemination have been described in the literature.
 Frequency
Frequency
Pleomorphic xanthoastrocytoma is a very rare neoplasm (< 1% of astrocytomas).
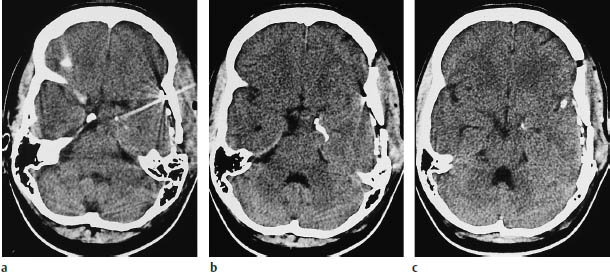
Fig.5.19a–c Pleomorphic xanthoastrocytoma. These CT scans were obtained after the placement of subdural electrodes.
a The scan at the basal level shows a mass lesion at the mesial left temporal pole.
b, c Scans at higher levels show linear calcification that is not specific for pleomorphic xanthoastrocytoma.
 Clinical Manifestations
Clinical Manifestations
 CT Morphology
CT Morphology
Pleomorphic xanthoastrocytoma is characterized mainly by its location. Arising from the cortex and leptomeninges, it shows a peripheral location on CT scans. It appears on noncontrast scans as a hypodense mass with sharp or indistinct margins. Most of these tumors enhance after contrast administration. Peripheral cysts are common, and calcifications are rare (Fig. 5.19). The tumors are always supratentorial and usually occur in the temporal lobe. Perifocal edema is often present.
Despite the characteristic meningeal component, the detection of a dural tail on MRI is not typical of pleomorphic xanthoastrocytoma.
 Differential Diagnosis
Differential Diagnosis
Differentiation is required from other tumors that abut the convexity and may infiltrate the dura. Pleomorphic xanthoastrocytoma is often misinterpreted as a glioblastoma or meningioma.
Subependymal Giant-Cell Astrocytoma
Frequency: subependymal nodules are present in almost all tuberous sclerosis patients (98% in some series), but subependymal giant-cell astrocytomas develop in only about 7–23%.
Suggestive morphologic findings: subependymal enhancing mass located at the foramen of Monro.
Procedure: obtain delineation of subependymal nodules (contrast administration).
Other studies: MRI is superior for distinguishing subependymal tumors from heterotopic tissue (isointense to cortex in all sequences).
Checklist for scan interpretation:
 Number, location, size of tumors?
Number, location, size of tumors?
 Contrast enhancement?
Contrast enhancement?
 Signs of infiltrative growth?
Signs of infiltrative growth?
 Other tumors or abnormalities?
Other tumors or abnormalities?
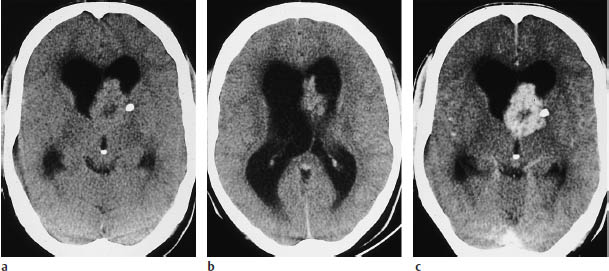
Fig. 5.20a–c Subependymal giant-cell astrocytoma. CT demonstrates enlargement of the lateral ventricles due to obstruction of the foramen of Monro.
a, b Even without contrast administration, nodular masses can be seen in the ventricular wall. A small calcification is visible on the left side.
c The mass enhances intensely after contrast administration. Presumed infiltration of the basal ganglia was confirmed histologically.
 Pathogenesis
Pathogenesis
Subependymal giant-cell astrocytomas (Fig. 5.20) are observed in tuberous sclerosis, one of the phakomatoses. They develop individually from subependymal tubers.
Subependymal giant-cell astrocytoma is an intermediate entity between heterotopia and neoplasia. Histologically, it does not exhibit mitoses or endothelial proliferation.
 Frequency
Frequency
Tuberous sclerosis has a reported annual incidence of approximately one per 178,000. Its prevalence is 10.6 per 100,000 population. Subependymal giant-cell astrocytomas develop in 7–23% of tuberous sclerosis patients, and in some cases may be the only detectable feature of the disease.
 Clinical Manifestations
Clinical Manifestations
Subependymal giant-cell astrocytomas are usually benign and slow-growing. Clinical symptoms are caused chiefly by the mass effect on the foramen of Monro, leading to obstructive hydrocephalus.
 CT Morphology
CT Morphology
While subependymal nodules in patients with tuberous sclerosis do not show contrast enhancement, subependymal giant-cell astrocytomas enhance intensely after contrast administration. Serial observations published by one group of authors suggest that the onset of contrast enhancement signals the transformation of a subependymal tuber into a giant-cell astrocytoma. Both lesions may contain calcifications, and both may be hyperdense to the white matter.
 Differential Diagnosis
Differential Diagnosis
The same diagnoses should be considered as with other subependymal masses:
• Ependymoma
• Subependymoma
• Heterotopic tissue
It is important to confirm or exclude tuberous sclerosis, which may present with the classic triad of adenoma sebaceum, seizures, and mental retardation.
 Follow-Up
Follow-Up
Subependymal giant-cell astrocytoma is rarely treated surgically, and there have been no reports on its propensity for recurrence.
Ependymal Tumors
Ependymoma
Frequency: makes up 2–8% of primary intracranial tumors, 15% of infratentorial posterior fossa tumors. In children, there appears to be a slight male predilection.
Suggestive morphologic findings: relation to ventricular ependyma, intense contrast enhancement, frequent calcification.
Procedure: only histologic examination can differentiate ependymoma from other tumors of ventricular origin. Seeding of CSF pathways may occur (spinal evaluation by MRI or myelography).
Other studies: the relation to the ventricle is often appreciated better on coronal or sagittal images (reformatted thin-slice CT or MRI).
Checklist for scan interpretation:
 Location?
Location?
 Hydrocephalus?
Hydrocephalus?
 Evidence of subarachnoid seeding?
Evidence of subarachnoid seeding?
Ependymomas arise from the ependyma lining the CSF spaces and may have a supratentorial, infratentorial, or spinal location. Infratentorial ependymoma (Figs. 5.21, 5.22) is common and mainly affects children under 4 years of age.
Supratentorial and spinal ependymomas are more common in young to middle-aged adults. The more benign forms grow by expansion and have well-defined margins on images; indistinct margins suggest a more malignant, infiltrative form. The potential for subarachnoid seeding to the spine necessitates adjunctive examinations of the spinal canal.
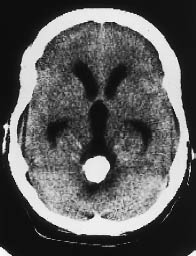
Fig. 5.21 Ependymoma. CT demonstrates a heavily calcified mass in the fourth ventricle, causing hydrocephalus.
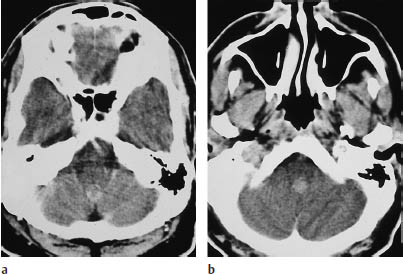
Fig. 5.22a, b Ependymoma. Both postcontrast scans show a moderately enhancing infratentorial mass approximately 1 cm in diameter. The tumor is located in the fourth ventricle.
 Pathogenesis
Pathogenesis
Ependymomas are classified as gliomas. The neuroepithelial forms arise from the ependymal lining of the CSF spaces. Mixed forms may occur. Intraparenchymal (ectopic) forms are usually supratentorial and arise from heterotopic ependymal cells.
The biologic behavior of ependymomas is variable, like that of other glial tumors. A highly malignant form is ependymoblastoma. The recurrence rate is relatively high, especially with infratentorial ependymomas that have invaded the cerebellopontine subarachnoid spaces. These tumors are difficult to resect completely because they tend to encase local blood vessels and cranial nerves (VII, IX, X). The potential for subarachnoid seeding also contributes to the generally poor prognosis.
 Frequency
Frequency
Ependymomas make up approximately 5–6% of intracranial gliomas, and 69% occur in children.
 Clinical Manifestations
Clinical Manifestations
As with other posterior fossa tumors, the clinical hallmarks are nausea, vomiting, and gait disturbance.
 CT Morphology
CT Morphology
Supratentorial ependymomas may show intraventricular and extraventricular growth. Their density is highly variable on unenhanced scans, and the tumors may appear isodense, hypodense, or hyperdense to the white matter. They enhance after contrast administration (Fig. 5.22). Approximately half of the tumors contain calcifications, and many supratentorial ependymomas are partly or completely cystic. Supratentorial ventricular expansion may occur, depending on the tumor location. Tumors that are completely or predominantly extraventricular can lead to eventual hydrocephalus (Figs. 5.21, 5.22), by which time the tumor has often reached a considerable size (5–8 cm in diameter).
Infratentorial ependymomas generally arise from the floor of the fourth ventricle, the expanded walls of which form a hypodense halo. Hydrocephalus is invariably present at diagnosis. Authors consistently emphasize the “plasticity” of these tumors, or their tendency to squeeze through the foramina of the fourth ventricle and spread into the subarachnoid space of the posterior fossa. The tumors may extend through the foramen of Luschka into the cerebellopontine angle cisterns, or through the foramen of Magendie to the spinal subarachnoid space. Tumors located on the floor of the fourth ventricle tend to expand the superior portion of the ventricle. Like their supratentorial counterparts, approximately 50% of infratentorial ependymomas contain calcifications. This leaves a sizable number that show little or no density contrast to their surroundings on unenhanced scans, but ependymomas invariably show marked enhancement after contrast administration. Foci of intratumoral hemorrhage may be observed.
 Differential Diagnosis
Differential Diagnosis
Infratentorial ependymoma in the pediatric age group requires differentiation from medulloblastoma (the most common posterior fossa tumor in children), from astrocytomas of the brain stem and cerebellum, and from rare tumors (ganglioglioma, hemangioblastoma, teratoma). Calcification is less common in medulloblastomas than in ependymomas. Medulloblastomas often show mixed density on plain CT scans due to intratumoral hemorrhage and necrosis. Ependymomas tend to show more conspicuous contrast enhancement.
The presence of cystic components helps to differentiate supratentorial ependymomas from other supratentorial tumors such as choroid plexus papilloma and meningioma. It is difficult to distinguish extraventricular ependymoma from other extraventricular tumors at a similar location, since any tumors in the glioma group may contain cysts and necrotic components.
 Follow-Up
Follow-Up
The standard criteria for the radiologic detection of recurrence are a reduction in the postoperative tissue defect, the appearance of new enhancing soft tissue, or progressive enlargement of enhancing tissue. Seeding into the subarachnoid space (including the spinal canal) may also be observed.
Subependymoma
Frequency: rare.
Suggestive morphologic findings: a solid mass in the ventricular wall, heterogeneous density, hyperdense with intratumoral hemorrhage, usually shows marked contrast enhancement. Calcification may be present.
Procedure: histologic evaluation is required, as subependymoma is not distinguishable from ependymoma by noninvasive methods.
Other studies: only MRI or myelography is suitable for spinal screening.
Checklist for scan interpretation:
 Location?
Location?
 Hydrocephalus?
Hydrocephalus?
 Evidence of subarachnoid seeding?
Evidence of subarachnoid seeding?
 Pathogenesis
Pathogenesis
As a rule, subependymomas are benign tumors that rarely become symptomatic. They are detected incidentally in CT examinations performed for some other indication (or at autopsy). Invasive growth and subependymal spread are unusual.
 Frequency
Frequency
Subependymoma is a rare tumor that is usually asymptomatic and is mainly discovered at autopsy.
 Clinical Manifestations
Clinical Manifestations
Supratentorial subependymomas generally do not cause clinical symptoms. It should be noted, however, that an absence of symptoms is mainly characteristic of supratentorial tumors. Infratentorial and spinal subependymomas lead to hydrocephalus or compression syndromes, even when biologically benign. Middle-aged and elderly adults are most commonly affected.
 CT Morphology
CT Morphology
Subependymomas form nodular masses of variable size in the ventricular wall. The tumor nodules are usually easy to distinguish from brain tissue and tend to have a small area of contact with it. Subependymomas may be hypodense or hyperdense. Extensive cystic change and extensive calcification appear to be unusual, although minute calcifications have been observed along with foci of intratumoral hemorrhage. Both changes may appear as hyperdense areas. Intense contrast enhancement does not rule out a diagnosis of subependymoma.
 Differential Diagnosis
Differential Diagnosis
The differential diagnosis includes ependymoma, which is almost indistinguishable from subependymoma by its CT features. Subependymal giant-cell astrocytoma can be identified by noting the cerebral stigmata of tuberous sclerosis or other cerebral changes that occur in this phakomatosis. Nodular masses in the ventricular wall may also consist of heterotopic cortical tissue. Because this tissue represents periventricular gray matter located within the white matter, it is equivalent to gray matter in its unenhanced CT density, enhancement characteristics, and signal intensity on MRI.
Choroid Plexus Papilloma and Carcinoma
Frequency: generally rare, making up approximately 0.5% of intracranial tumors in adults. Accounts for 10% of intracranial tumors before 1 year of age, however, and 3–5% of intracranial tumors in children.
Suggestive morphologic findings: “fern-like” pattern, intense enhancement.
Procedure: coronal reformatted images can be helpful for differentiating choroid plexus tumor from secondary intraventricular tumor. Contrast administration is always indicated.
Other studies: MRI is advantageous owing to arbitrary plane selection, and is useful for general screening of the neuraxis for CSF metastases.
Checklist for scan interpretation:
 Location?
Location?
 Hydrocephalus?
Hydrocephalus?
 Pathogenesis
Pathogenesis
Choroid plexus carcinoma is a highly malignant variant of choroid plexus papilloma. Infiltration of the white matter with extensive perifocal edema should raise suspicion of the malignant variant, which also causes seeding of CSF pathways. Choroid plexus papilloma is most common before 2 years of age but may also occur in adults.
 Frequency
Frequency
On the whole, choroid plexus tumors are rare. They make up approximately 0.4-1% of intracranial tumors, depending on the source. About 70% of patients are under 2 years of age.
 Clinical Manifestations
Clinical Manifestations
A large percentage of choroid plexus papillomas are diagnosed in the first year of life, although the tumor may occur in later decades. The lateral ventricles are more commonly affected in children, and the fourth ventricle in adults. Males predominate. Severe hydrocephalus is almost always present and results from excessive CSF production by the tumor rather than obstruction.
 CT Morphology
CT Morphology
The tumors typically show an irregular, “fernlike” surface. Supratentorial tumors are often located in the trigone of the lateral ventricle, and infratentorial tumors are often found in the fourth ventricle (Fig. 5.23).
Choroid plexus tumors are isodense or slightly hyperdense to the white matter on noncontrast scans. Calcifications are frequent. Intense enhancement occurs after contrast administration. Papillomas are indistinguishable from carcinoma in the absence of infiltrative growth. Multiple tumors are occasionally observed.
 Differential Diagnosis
Differential Diagnosis
Because of their intense contrast enhancement, at least on CT scans, vascular malformations of the choroid plexus are apt to be confused with choroid plexus papilloma. They can be differentiated by unenhanced MRI (vascular flow void) or by angiography. Cysts of the choroid plexus are a more common finding. They are generally asymptomatic, but they may cause obstruction, and are occasionally mistaken for tumors. Cysts may be unilateral or bilateral. Older studies attributed them to cystic degeneration of the choroid plexus, but the same lesions have been described in children. Carcinoma can develop as a malignant variant of choroid plexus papilloma, and may show an infiltrative growth pattern on CT.
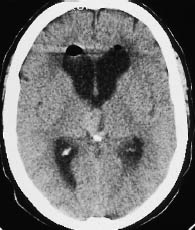
Fig. 5.23 Choroid plexus papilloma. Noncontrast CT demonstrates a hyperdense mass with smooth margins in the third ventricle, rostral to the calcified pineal gland.
Neuroepithelial Tumors of Unknown Origin
Gliomatosis Cerebri
Frequency: rare.
Suggestive morphologic findings: increase in brain volume, hypodensities.
Procedure: no causal treatment, stereotactic biopsy.
Other studies: diagnosis requires demonstrating multiple-lobe involvement, so it is insufficient to biopsy a single site.
Checklist for scan interpretation:
 Enough evidence to make a presumptive diagnosis?
Enough evidence to make a presumptive diagnosis?
 Pathogenesis
Pathogenesis
Gliomatosis cerebri is distinguished from primary brain tumors; this applies to unifocal gliomas, as well as multifocal and multicentric forms. Gliomatosis cerebri involves a diffuse infiltration of the brain by neoplastic astrocytes, often occurring along anatomic or functional structural units. White-matter tracts are predominantly affected. Brain anatomy is preserved.
Gliomatosis cerebri is slowly progressive, but published reports include cases with a peracute course as well as indolent cases that apparently develop over decades.
 Frequency
Frequency
Gliomatosis cerebri is a very rare disease. Accurate data have not been published regarding its incidence or prevalence.
 Clinical Manifestations
Clinical Manifestations
Patients exhibit nonspecific personality changes.
 CT Morphology
CT Morphology
CT shows a substantial increase in brain volume accompanied by more or less diffuse hypodensity. The ventricles are correspondingly small, unless there has been outflow obstruction leading to localized expansion. There have been isolated reports of solitary or multiple hypodense foci that may show contrast enhancement.
 Differential Diagnosis
Differential Diagnosis
A definitive diagnosis by brain biopsy is usually preceded by several misdiagnoses. An infiltrative glioma may be mistaken for gliomatosis cerebri, but generally a focal mass lesion can be identified somewhere in the brain. It is very difficult to distinguish gliomatosis cerebri from leukoencephalopathy, multifocal leukoencephalopathy, multiple sclerosis, and various ischemic changes. A helpful criterion is that leukoencephalopathy, unlike gliomatosis, never involves the gray matter.
Neuronal and Mixed Neuronal/Glial Tumors
Gangliocytoma
Frequency: very rare.
Suggestive morphologic findings: cystic and calcified tumor-like mass.
Procedure: MRI.
Other studies: MRI is more sensitive than CT (no beam-hardening artifact, Fig. 5.24 b).
Checklist for scan interpretation:
 Calcifications, cysts, enhancement characteristics?
Calcifications, cysts, enhancement characteristics?
 Compression of fourth ventricle? Hydrocephalus?
Compression of fourth ventricle? Hydrocephalus?
 Pathogenesis
Pathogenesis
Gangliocytoma is a low-grade nerve cell tumor. A more malignant variant is ganglioglioma. Lhermitte-Duclos disease is a variant affecting the cerebellar hemispheres.
 Frequency
Frequency
Extremely rare.
 Clinical Manifestations
Clinical Manifestations
Gangliocytoma is often located in the temporal lobe, where it typically produces seizures. Occurrence in the posterior fossa leads to cerebellar symptoms such as gait disturbance and vertigo. More extensive spread leads to cranial nerve deficits.
 CT Morphology
CT Morphology
Cystic and calcified structures are a typical finding. There is usually no contrast enhancement.
 Differential Diagnosis
Differential Diagnosis
The main tumor to be considered in differential diagnosis is oligodendroglioma. Chondrosarcoma is a rare tumor that may present with similar features.
Ganglioglioma
Frequency: 0.3–0.9% of brain tumors; more common in children and adolescents (up to 8%).
Suggestive morphologic findings: hypodense (cystic) temporal mass, often with calcification or rim enhancement.
Procedure: temporal lobe CT projection.
Other studies: MRI can demonstrate temporal lobe tumors without artifacts.
Checklist for scan interpretation: the diagnosis is rarely made prior to surgery. The key points are as follows:
 Location?
Location?
 Structure: cystic, solid, calcifications?
Structure: cystic, solid, calcifications?
 Mass effect, edema?
Mass effect, edema?
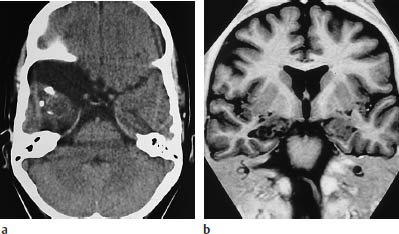
Fig. 5.24a, b Gangliocytoma. The magnetic resonance imaging (MRI) sequence, chosen to optimize contrast between the white matter and the cortex, clearly demonstrates a rounded mass in the right mesial temporal lobe.
a CT clearly demonstrates the partially calcified mass. In addition, there is an arachnoid cyst adjacent to the temporal pole.
b MRI. The tumor is more clearly visualized.
 Pathogenesis
Pathogenesis
 Frequency
Frequency
Ganglioglioma, like gangliocytoma, is extremely rare. It mainly affects children and young adults. The peak incidence is at approximately age 11.
 Clinical Manifestations
Clinical Manifestations
Temporal lobe epilepsy is a common presenting feature in patients with ganglioglioma. Increasing frequency of seizures is an important clue to the underlying tumor. A long history of headaches is often present.
 CT Morphology
CT Morphology
The CT appearance of gangliogliomas is somewhat variable. They are usually supratentorial, showing a preference for the temporal lobes. An infratentorial location is less characteristic. One author, however, cites the floor of the third ventricle as a typical site of occurrence. The tumor is hypodense on noncontrast CT, and often contains one or more cysts, which appear as foci of water density. Calcifications are found in approximately one-third of gangliogliomas (Fig. 5.25).
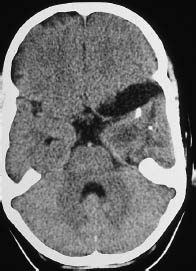
Fig. 5.25 Ganglioglioma. Noncontrast CT in a 10-year-old girl shows expansion of the extra-axial cerebrospinal fluid spaces over the left temporal pole, accompanied by an intraparenchymal, hypodense mass approximately 2 cm in diameter, containing coarse calcifications.
Contrast enhancement of the solid portion of the tumor occurs in about half of the cases.
 Differential Diagnosis
Differential Diagnosis
Ganglioglioma may be omitted from the differential diagnosis because of its rarity, but should be considered in the differential diagnosis of infratentorial tumors, along with Lhermitte-Duclos disease, a hamartoma-like dysplasia.
 Follow-Up
Follow-Up
The treatment of choice is surgical removal. Although the tumors appear to have well-defined margins, complete removal may not be possible, depending on the location. However, even an incomplete resection may be followed by good long-term survival, with clinical improvement and no significant tumor growth. Consequently, postoperative radiation and chemotherapy are usually withheld.
Central Neurocytoma
Frequency: rare tumor, frequently mistaken for oligodendroglioma.
Suggestive morphologic findings: purely intraventricular mass with cysts and calcifications in young adults.
Procedure: a presumptive diagnosis can be made from CT findings.
Other studies: MRI is less rewarding, as it cannot confirm calcification. The angiographic findings are nonspecific.
Checklist for scan interpretation:
 Strictly intraventricular location?
Strictly intraventricular location?
 Calcifications?
Calcifications?
 Pathogenesis
Pathogenesis
While neuroblastoma can be viewed as a more malignant type of neuroectodermal tumor, central neurocytoma is a benign but related variant of the latter type of lesion.
 Frequency
Frequency
Central neurocytoma has not been accepted as a separate entity for very long, and accurate data on its incidence and prevalence are therefore not yet available. On the whole, however, the tumor is rare.
 Clinical Manifestations
Clinical Manifestations
Clinical symptoms are nonspecific. They include headache and symptoms caused by hydrocephalus.
 CT Morphology
CT Morphology
CT in typical cases demonstrates an intraventricular mass with moderately well-defined margins, mild to moderate contrast enhancement, and coarse calcifications. Multiple small cysts are also common. The tumor is isodense or slightly hypodense to the white matter on noncontrast scans. A frequent location is near the foramen of Monro, leading to expansion of the lateral ventricles. At least one case of noninvasive tumor extension through the third ventricle into the fourth ventricle has been described in the literature.
 Differential Diagnosis
Differential Diagnosis
Among tumors that have a subependymal origin, ependymomas and subependymomas are typically located in the fourth ventricle. Ependymomas occur predominantly in children, while neurocytoma is a tumor of young adults. The differential diagnosis should include intraventricular gliomas, which are distinguished by edema of the peritumoral white matter. Choroid plexus papillomas enhance intensely after contrast administration, and occur mainly in children.
 Follow-Up
Follow-Up
With regard to the problem of residual or recurrent tumor, published studies indicate that even incomplete tumor removal leads to a regression of symptoms.
Esthesioneuroblastoma
Frequency: rare, but probably underdiagnosed.
Suggestive morphologic findings: mass below the cribriform plate.
Procedure: surgical removal.
Other studies: coronal MRI is useful for detecting intracranial extension.
Checklist for scan interpretation:
 Destruction of cribriform plate?
Destruction of cribriform plate?
 Intracranial extension?
Intracranial extension?
 What spaces are affected (paranasal sinuses, etc.)?
What spaces are affected (paranasal sinuses, etc.)?
 Tumor stage according to Kadish et al. (1976) (see below).
Tumor stage according to Kadish et al. (1976) (see below).
 Pathogenesis
Pathogenesis
Esthesioneuroblastoma arises from olfactory epithelial cells. Tumors such as neuroblastoma, pheochromocytoma, and the forms occurring in multiple endocrine neoplasia (MEN) syndromes have a related origin.
The following pretherapy staging system can be used:
• Stage A: tumor is limited to the nasal cavity.
• Stage B: tumor is localized to the nasal cavity and one or more paranasal sinuses.
• Stage C: tumor has infiltrated the orbits, skull base, or cervical lymph nodes, or there is other metastatic spread.
Therapeutic approaches vary. Given the relatively poor prognosis, surgical treatment is generally supplemented by radiotherapy, chemotherapy, or both.
 Frequency
Frequency
 Clinical Manifestations
Clinical Manifestations
Reports on affected age groups vary in different series. The case numbers are probably too small for reliable estimates. A slight male predominance has been noted in some series.
Symptoms result from the rich blood supply of the tumor (often supplied by the ophthalmic artery) and its location. Epistaxis and nasal airway obstruction are observed.
 CT Morphology
CT Morphology
Direct coronal CT scans (plain and postcontrast, soft-tissue window, bone window) are recommended (Fig. 5.26).
The typical site of occurrence is the upper nasal cavity, below the cribriform plate. The tumors display a very homogeneous density before and after contrast administration. Intratumoral hemorrhage and cysts are usually absent. Calcifications are common, but bone destruction is also common, making it difficult to distinguish tumoral calcification from displaced bone fragments. Erosive bone changes suggest extension into adjacent compartments (Fig. 5.26).
 Differential Diagnosis
Differential Diagnosis
The radiologic differential diagnosis of esthesioneuroblastoma is difficult. The tumor should be considered in the evaluation of all midline masses that appear to be located in the anterior fossa. Differentiation is particularly required from frontobasal meningioma, which may show extracranial extension into the paranasal sinuses with a midline location. Bone-forming tumors, chondrosarcoma, and similar entities are less homogeneous, consistently calcify, and show less contrast enhancement. Fibrous dysplasia also has a nonhomogeneous appearance. Squamous-cell carcinoma is the most common tumor that can mimic esthesioneuroblastoma in its imaging morphology.
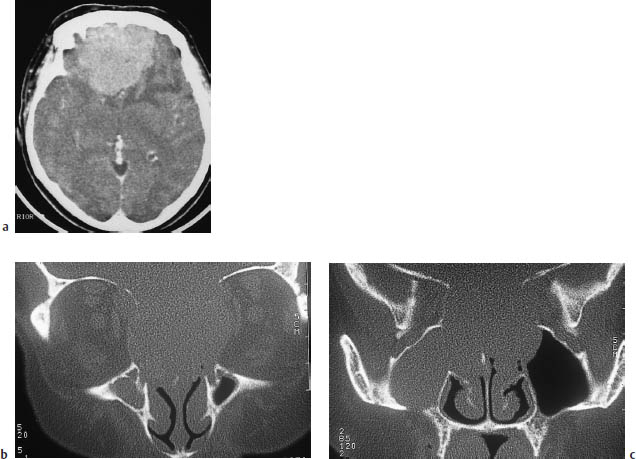
Fig. 5.26a–c Esthesioneuroblastoma.
a An axial CT scan after contrast administration shows a very large mass with perifocal edema, abutting the anterior skull base.
b, c On direct coronal scans with a bone window, the very large extracerebral tumor component filling the ethmoid cells and sphenoid sinus and extending into the left frontal sinus helps to distinguish the tumor from meningioma.
Pineal Parenchymal Tumors
Pineocytoma, Pineoblastoma, Pineal Cyst
Frequency: rare; pineocytomas and pineoblastomas are considerably rarer than germinomas.
Suggestive morphologic findings: mass arising from the pineal region.
Procedure: MRI may be required. Contrast administration is essential.
Other studies: sagittal MRI contributes to accurate localization.
Checklist for scan interpretation:
 Hydrocephalus?
Hydrocephalus?
 Location in relation to third ventricle, vein of Galen, quadrigeminal plate?
Location in relation to third ventricle, vein of Galen, quadrigeminal plate?
 Calcifications, signs of infiltrative growth?
Calcifications, signs of infiltrative growth?
 Pathogenesis
Pathogenesis
Pineocytoma is the more benign type of pineal parenchymal tumor. The more aggressive form is pineoblastoma.
 Frequency
Frequency
Pineal region tumors are relatively rare, making up approximately 3–8% of brain tumors in children and less than 1% in adults. Pineal cysts, by contrast, are a common incidental finding. In one large series, they were detected in about 4% of MRI examinations, and they are found in some 25–40% of autopsies. The frequency distribution of pineal region tumors in a series of 370 cases was as follows:
• Germinomas 27%
• Astrocytomas 26%
• Pineocytomas 12%
• Pineoblastomas 12%
• Ependymomas 4%
• Other tumors, including lymphomas and metastases, < 3%
 Clinical Manifestations
Clinical Manifestations
Nearly all patients with pineal region tumors become symptomatic due to impairment of CSF circulation. The manifestations may be headache, vomiting, lethargy, or increased head circumference in children. Hormone-producing tumors can lead to precocious puberty and other endocrinopathies, depending on their specific substrates. A mass effect on the quadrigeminal plate may lead to Parinaud’s syndrome.
 CT Morphology
CT Morphology
Published reports vary on the CT features of these rare tumors. They are usually described as appearing isodense or hyperdense to normal brain tissue and as showing moderate to marked contrast enhancement. Pineoblastomas show early infiltrative growth, while pineocytomas are more sharply marginated. Calcifications are found in pineocytoma, but less consistently than in germinoma. Calcifications apparently do not occur in pineoblastoma.
 Differential Diagnosis
Differential Diagnosis
An aggressive pineoblastoma that infiltrates the cerebellar vermis may be mistaken for medulloblastoma.
Medulloepithelioma
Frequency: very rare.
Suggestive morphologic findings: smoothly marginated cystic/solid mass.
Procedure: radical excision.
Other studies: multiplanar MRI.
Checklist for scan interpretation:
 Contrast enhancement (atypical)?
Contrast enhancement (atypical)?
 Smooth margins (typical)?
Smooth margins (typical)?
 Intracranial or spinal metastases via CSF pathways?
Intracranial or spinal metastases via CSF pathways?
 Pathogenesis
Pathogenesis
Medulloepithelioma is included in the class of primitive neuroectodermal tumors (PNETs). It bears a close histologic resemblance to the primitive cells of the neural tube. Medulloepitheliomas are highly malignant, and survival is often less than one year. While cerebral medulloepithelioma has a poor prognosis, the ocular form of the tumor takes a much more favorable course. Children with ocular medulloepithelioma often remain disease-free after enucleation of the eye.
 Frequency
Frequency
Medulloepithelioma is extremely rare.
 Clinical Manifestations
Clinical Manifestations
Medulloepithelioma is a tumor of early childhood. Most cases have been described during the first years of life, and occurrence after age 10 is very unusual.
 CT Morphology
CT Morphology
Medulloepitheliomas are usually located in the periventricular white matter. Occurrence in the brain stem has also been reported. Like other PNETs, they are sharply marginated. Generally they have both cystic and solid components. They are isodense or hypodense to surrounding tissues on noncontrast CT scans and usually do not enhance after contrast administration. Intratumoral hemorrhage is occasionally observed, but calcifications are absent.
 Differential Diagnosis
Differential Diagnosis
Medulloepitheliomas located in the brain stem require differentiation from gliomas. This form is distinguished from pilocytic astrocytoma by its lack of enhancement after contrast administration and by its well-defined margins.
 Follow-Up
Follow-Up
Like other intracerebral tumors, medulloepitheliomas have a tendency to disseminate via CSF pathways. Consequently, the radiologist should look for intracranial and spinal metastases during the initial examination and in follow-ups.
Neuroblastoma
Frequency: the primary intracerebral form is rare. Metastases are more common.
Suggestive morphologic findings: supratentorial mass, frequently large, containing calcifications, hemorrhagic foci, and cysts.
Procedure: seeding via CSF pathways, including the spinal canal, must be investigated.
Other studies: MRI is superior for detecting CSF dissemination, particularly to the spine.
Checklist for scan interpretation:
 Typical combination of intratumoral hemorrhage, calcifications, and cysts?
Typical combination of intratumoral hemorrhage, calcifications, and cysts?
 Infiltration of dural sinuses (risk of distant metastasis with need to evaluate chest, etc.)?
Infiltration of dural sinuses (risk of distant metastasis with need to evaluate chest, etc.)?
 Relation to ventricles (CSF dissemination)?
Relation to ventricles (CSF dissemination)?
 Hydrocephalus?
Hydrocephalus?
 Note necessity for MRI evaluation (for spinal metastases).
Note necessity for MRI evaluation (for spinal metastases).
 Pathogenesis
Pathogenesis
Neuroblastomas are highly malignant tumors, which frequently recur and are associated with a high incidence of CSF and non-CSF metastases. The precise histologic classification of neuroblastomas is controversial.
 Frequency
Frequency
Very rare.
 Clinical Manifestations
Clinical Manifestations
Clinical symptoms vary with the site of occurrence. General signs of increased intracranial pressure are most commonly observed. Seizures and hemiparetic symptoms can also occur.
 CT Morphology
CT Morphology
The CT appearance of neuroblastoma (Fig. 5.27) is fairly characteristic.
The tumors are predominantly supratentorial and often show a relation to the ventricular system. Spontaneous intratumoral hemorrhage often leads to a patchy increase in density on unenhanced scans. Calcifications are common, as are cystic components of variable size. Marked enhancement occurs after contrast administration.
CSF dissemination is a frequent and specific feature of neuroblastoma. Nodular metastases may be found in the ventricles or extra-axial CSF spaces, and there may be tumor encasement of the spinal cord.
 Differential Diagnosis
Differential Diagnosis
A heterogeneous appearance is characteristic of various other tumors. Oligodendrogliomas, especially the more malignant forms, can have a variety of imaging features. This is also true of astrocytomas in children. Ependymomas and other PNETs should also be included in the differential diagnosis.
 Follow-Up
Follow-Up
Recurrences are common, and 5-year survival rates are often low even with comprehensive treatment.
Ependymoblastoma
This very rare tumor is included in the group of neuroblastomas, PNETs, and medulloblastomas in the WHO classification. It is a highly malignant, usually intraventricular tumor, with infiltrating margins and a poor prognosis. Ependymoblastoma cannot be reliably distinguished from other intraventricular tumors by CT.
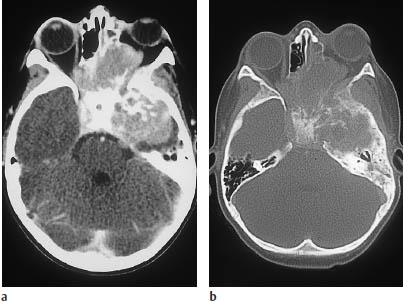
Fig. 5.27a, b Metastatic neuroblastoma in a 12-year-old child.
a Postcontrast CT shows an extensive mass involving the middle fossa and paranasal sinuses, with invasion of the left orbit.
The bone-window scan reveals extensive bone destruction.
Primitive Neuroectodermal Tumor/ Medulloblastoma
Frequency: approximately every fifth or sixth brain tumor in children is a medulloblastoma, and it also accounts for one-third of posterior fossa tumors in the pediatric age group.
Suggestive morphologic findings: rounded midline mass, hyperdense on noncontrast CT, occurring in a child. Often located in the roof of the fourth ventricle (fastigium).
Procedure: spinal and cerebral MRI (to detect metastatic deposits).
Other studies: MRI is more sensitive than CT for detecting metastatic deposits and tumor recurrence (posterior fossa).
Checklist for scan interpretation:
 Location (midline/hemispheric, infratentorial)?
Location (midline/hemispheric, infratentorial)?
 Unenhanced density? Response to contrast administration?
Unenhanced density? Response to contrast administration?
 Calcifications, cysts, blood?
Calcifications, cysts, blood?
 Hydrocephalus?
Hydrocephalus?
 Tumor nodules located elsewhere in the brain or at the craniocervical junction?
Tumor nodules located elsewhere in the brain or at the craniocervical junction?
 Pathogenesis
Pathogenesis
Metastases from medulloblastoma are not uncommon and are seeded via CSF pathways. Accordingly, metastatic nodules may be found in the cisterns, spinal canal, and leptomeninges. Transpial spread can also occur via the Virchow-Robin perivascular spaces.
Medulloblastomas are classified as grade IV tumors and are among the most aggressive pediatric brain tumors.
 Frequency
Frequency
As a prototype PNET, medulloblastoma is the most common brain tumor in children. It accounts for approximately 20–25% of pediatric brain tumors, and has an incidence of 0.5 per 100000 children. Some 70% of medulloblastomas are diagnosed in patients under 16 years of age, and 80% of affected adults are between age 21 and 40. The peak occurrence is at about 7 years of age. Approximately 65% of patients are male. Seventy-five percent of medulloblastomas arise from the cerebellar vermis.
 Clinical Manifestations
Clinical Manifestations
Most patients are under 10 years of age, but a second peak occurs at the end of adolescence. Males predominate by a ratio of 4: 1.
Symptoms result from hydrocephalus or vertigo and nausea. Cranial nerve deficits are most often caused by tumors that extend laterally to involve the cerebellopontine cistern.
 CT Morphology
CT Morphology
CT examination in most patients demonstrates a round or elliptical, infratentorial midline mass. The tumor arises from the cerebellar vermis and displaces the fourth ventricle rostrally. A striking feature is its mild hyperdensity on noncontrast CT scans (Fig. 5.28).
Medulloblastomas typically show moderate to intense contrast enhancement. Secondary hydrocephalus is almost always present. Calcifications are unusual. The tumors often appear remarkably well-demarcated on images (and at surgery), but this is often found to be misleading on histologic examination. Tumors in the lateral cerebellum are more likely to occur in young adults, and can be difficult to classify in the differential diagnosis.
Less typical cases are distinguished by their nonhomogeneity on noncontrast CT and by irregular or minimal contrast enhancement. Calcifications were found in 17% of tumors in one large series, but intratumoral hemorrhage is extremely rare.
With regard to invasiveness, some correlation has been noted between the sharp margins seen on CT scans and the good demarcation of the tumor found at surgery.
 Differential Diagnosis
Differential Diagnosis
Astrocytomas are an important differential diagnosis. Unlike medulloblastomas, they usually appear hypodense on unenhanced scans. The presence of cysts does not exclude medulloblastoma, however. Similarly, calcifications do not favor a diagnosis of ependymoma over medulloblastoma, although it is important to note that medulloblastoma is a far more common tumor in children.
 Follow-Up
Follow-Up
Recurrent medulloblastoma resembles the primary tumor in that it also appears as a sharply marginated tumor nodule with homogeneous enhancement. Calcifications are more frequently described, however (56 %). CSF dissemination may occur even without a recurrent tumor. The metastases may form a “sheetlike” growth, or discrete nodular deposits on enhancing sulci and cisterns.
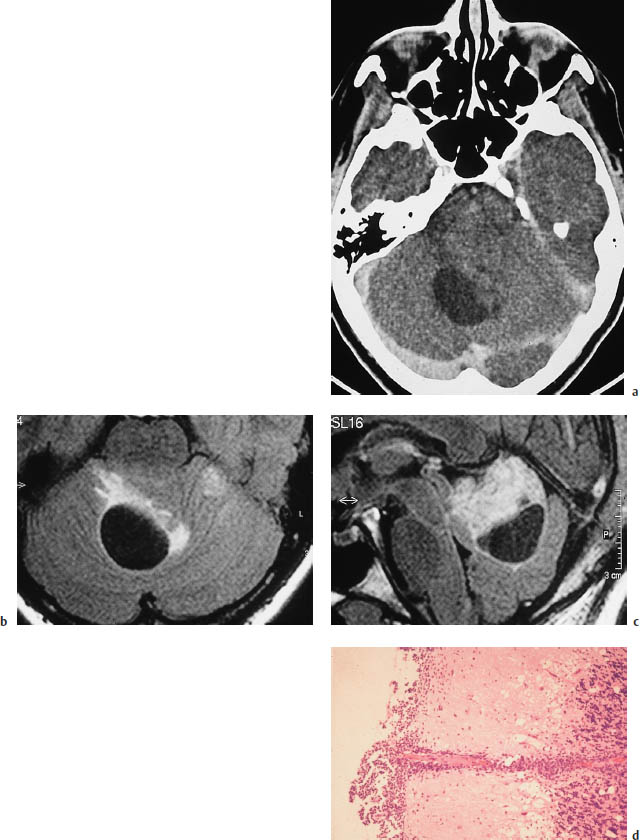
Fig.5.28a–d Medulloblastoma in a 30-year-old woman.
a Postcontrast CT demonstrates a large infratentorial tumor cyst. Nonenhancing soft tissue is visible to the left of the cyst.
b, c The tumor is defined most clearly by magnetic resonance imaging using a fluid-attenuated inversion recovery (FLAIR) sequence, with T2-weighted images in which the cerebrospinal fluid (CSF) appears hypointense. The sagittal image gives the best portrayal of tumor size.
d Histology. Leptomeningeal seeding with secondary infiltration of the cerebellum along the perivascular CSF spaces of the pial vessels.
Schwannoma (Neurinoma)
Frequency: one of the most common intracranial tumors. The incidence is higher in neurofibromatosis.
Suggestive morphologic findings: cerebellopontine angle mass expanding the internal acoustic meatus (bone window). May appear as an enhancing intrameatal mass.
Procedure: CT imaging of the posterior fossa with a bone window and 2-mm slice thickness is performed to demonstrate the jugular bulb in relation to the tumor (a “high bulb” poses an obstacle to surgery).
Other studies: MRI demonstrates cerebellopontine angle tumors better than CT, owing to the lack of beam-hardening artifact. This applies to the differential diagnosis and the detection of spread.
Checklist for scan interpretation:
 Check for high position of the jugular bulb (Fig. 5.29).
Check for high position of the jugular bulb (Fig. 5.29).
 Pathogenesis
Pathogenesis
Acoustic schwannomas may be unilateral or bilateral. Consistent with their clinical features, but contrary to their name, acoustic schwannomas arise from the vestibular nerve. The tumors are benign and require definitive surgical removal.
Some acoustic schwannomas occur in the setting of neurofibromatosis (Fig. 5.30). These tumors are often bilateral; they are diagnosed at an early age and coexist with meningiomas (bilateral acoustic schwannomas are pathognomonic for neurofibromatosis type 2).
Rarely, schwannomas occur within the brain parenchyma. With regard to the origin of these tumors, most authors agree that they arise from the Schwann cells of the perivascular neural plexus.
Another atypical site for schwannomas is the labyrinth. They can be detected on magnification views of the petrous bone, appearing as soft-tissue masses located near the foramen rotundum. Differentiation is required from glomus tympanicum tumors. Except for the optic nerve, which is devoid of Schwann cells, schwannomas can arise from any of the cranial nerves. They are relatively easy to diagnose when they have an extra-axial location and have caused the expansion of bony canals.
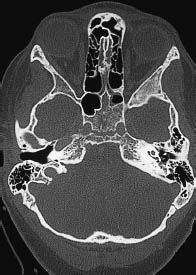
Fig. 5.29 High position of the right jugular bulb. The high jugular bulb appears as a rounded mass bounded by cortical bone in the posterior part of the petrous bone, contrasting with a normal appearance on the left side. The high bulb is situated within the surgical access route to the cerebellopontine angle. It should not be mistaken for an osteolytic lesion, associated with a glomus tumor, etc.
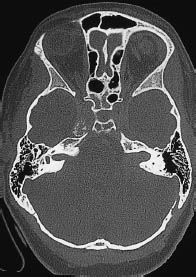
Fig. 5.30 Expansion of the internal auditory canal. Bilateral expansion of the internal acoustic meatus in bone-window CT is characteristic of acoustic neuromas in neurofibromatosis.
 Frequency
Frequency
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


