Chapter 11 Intraocular Tumors

Retinoblastoma
Retinoblastoma is the most common intraocular malignancy of childhood and occurs with a frequency of approximately one in 14,000 to 20,000 live births.1 Ninety percent of cases are diagnosed in children under the age of 3 years. Ultrasonography along with other forms of imaging is invaluable in establishing the diagnosis of retinoblastoma.
Clinical features, symptoms, and signs
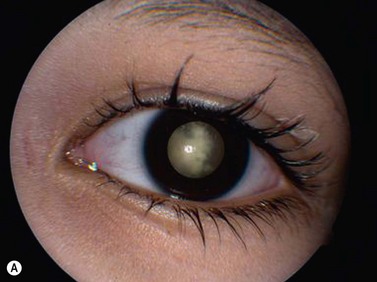
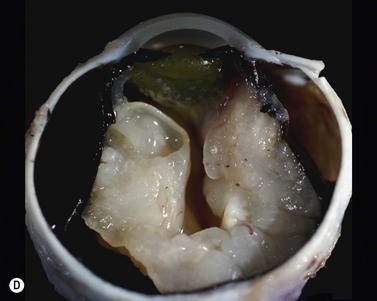
While leukocoria is the most common presenting symptom of retinoblastoma, strabismus, decreased vision, ocular inflammation, and other rarer symptoms have also been observed.1 In general, the presentation varies with the stage of the disease at the time of diagnosis. In its earliest clinical stage, retinoblastoma appears as a flat transparent to slightly whitish colored lesion in the sensory retina. Dilated and tortuous feeding retinal vessels may be evident. As the tumor enlarges, it loses its transparency and takes on a creamy yellow to whitish coloration with foci of chalk-like calcification. As it grows beyond the boundary of the sensory retina, retinoblastoma will typically follow either an endophytic or exophytic growth pattern (Figure 11.1). Other growth patterns including mixed and diffuse infiltrative forms (Figure 11.2) are less commonly observed. Necrosis may be a significant component of the tumor. Endophytic retinoblastomas grow from the retina inward towards the vitreous cavity. Vitreous seeding from these friable tumors as well as anterior chamber involvement can simulate endophthalmitis and other inflammatory conditions. In contrast, exophytic retinoblastomas grow from the retina outward into the subretinal space and can cause exudative retinal detachment, sometimes displacing the retina anteriorly behind the lens. Advanced retinoblastoma can present with neovascular glaucoma, corneal edema, spontaneous hyphema, vitreous hemorrhage, pseudohypopyon, and vitreitis.
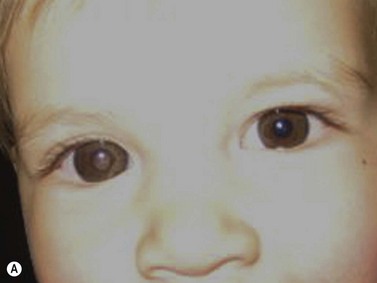
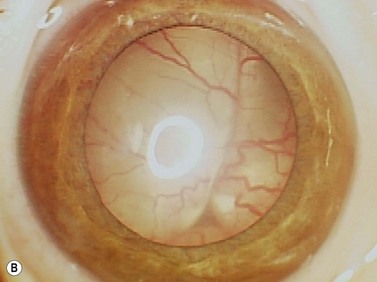
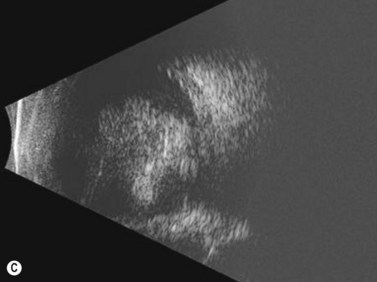
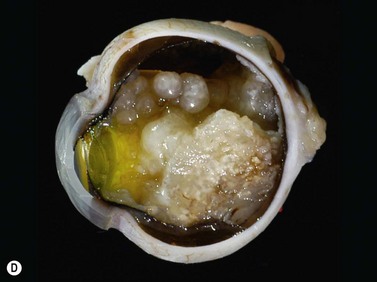
Diagnostic evaluation
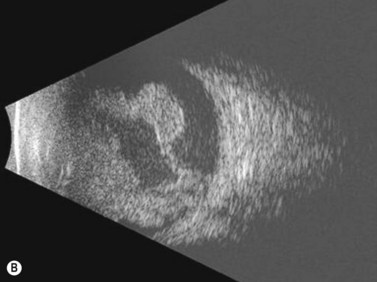
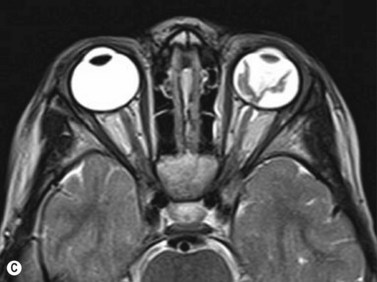
Ultrasonography is helpful in confirming the diagnosis of retinoblastoma and in differentiating the disease from other causes of leukocoria. This is particularly valuable when funduscopic examination is limited in advanced cases. On A-scan, the internal reflectivity of these lesions varies in accordance to the degree of calcification within the tumor. Non-calcified tumors exhibit low to medium internal reflectivity, whereas calcified lesions demonstrate high internal reflectivity. When a significant degree of calcification is present, shadowing of the adjacent sclera and orbit occurs. B-scan ultrasonography typically displays a rounded or irregular intraocular mass. It should be noted that mildly elevated and diffuse lesions have also been reported.2,3 Other associated ultrasonographic findings may include retinal detachment and vitreous opacities. When extraocular extension is present in cases of retinoblastoma, invasion of the optic nerve is the most common route. In cases where extensive calcification is present, tumor involvement of the optic nerve and extraocular extension can be difficult to detect secondary to the shadowing effect. CT and MRI imaging of the orbits should be used in combination with ultrasonography when optic nerve or extraocular invasion is suspected (Figure 11.2). MRI of the optic nerve, orbits, and brain is preferred as this modality offers superior soft tissue resolution and avoids potentially harmful exposure to radiation.
Salient diagnostic findings
The diagnosis of retinoblastoma can generally be suspected based upon the clinical findings observed in a complete ophthalmic examination in the office or an examination performed under anesthesia. The most commonly observed finding is an elevated intraocular mass with characteristic calcification demonstrating either an endophytic or exophytic growth pattern. Other causes of intraocular calcification are listed in Box 11.1.
Differential diagnosis
There are several pediatric ocular conditions that can cause leukocoria and should be considered in the differential diagnosis of retinoblastoma. The conditions that most commonly present a diagnostic challenge include retinopathy of prematurity (ROP), persistent fetal vasculature (PFV), Coats’ disease, toxocariasis, and medulloepithelioma (Table 11.1).
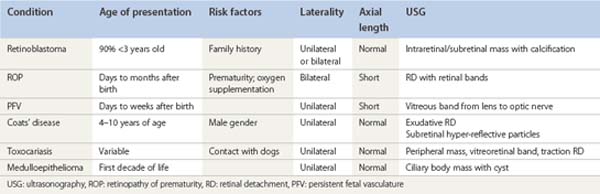
Retinopathy of prematurity
ROP occurs in the setting of known risk factors including: prematurity, low birth weight, and exposure to supplemental oxygenation in the neonatal period. While both ROP and retinoblastoma can present with leukocoria, in ROP the absence of the red reflex is caused by retinal dragging toward fibrovascular tissue in the retinal periphery. Eyes that develop retinoblastoma are usually of normal axial length. In contrast, in ROP it is more common for eyes to have some degree of the axial length shortening. Additionally, ROP is typically a bilateral condition whereas retinoblastoma can be either unilateral or bilateral. In the most advanced cases of ROP, the retina is detached in a funnel-like configuration, resulting in a hyper-reflective retrolental membrane on B-scan. The peripheral retina frequently exhibits a loop or trough-like appearance as a result of traction by the retrolental membrane (Figure 11.3).
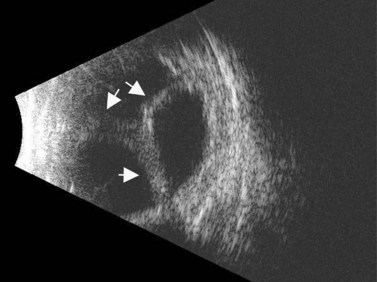
Persistent fetal vasculature
The lens is often thin with irregularities in the posterior capsule (Figure 11.4). Eyes usually have some degree of axial length shortening. Calcification may be present, however in contrast to retinoblastoma, there is no discrete mass visualized clinically or with ultrasonography.
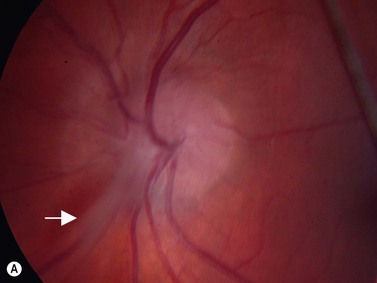
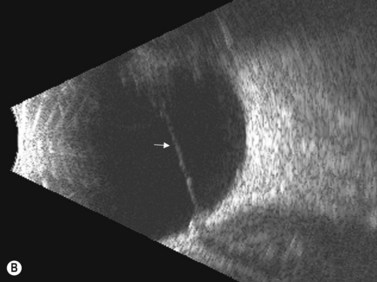
Coats’ disease
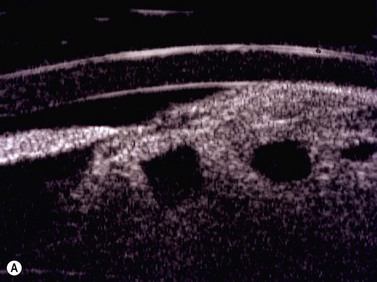
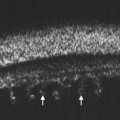
Coats’ disease is a retinal vascular disorder characterized by telangiectasia, intraretinal exudation, and exudative retinal detachment. Although Coats’ disease can present at any age, it usually is diagnosed in young males between 4 and 10 years of age.4 It is most commonly a unilateral disease process. In the early stages of Coats’ disease, localized, shallow retinal detachments may occur. In more advanced cases, total exudative detachments secondary to leakage from aneurysmal blood vessels are observed. This exudative process results in yellow cholesterol crystal deposition in the subretinal space that can be observed clinically as refractile bodies. These particles are much less reflective than the calcium particles in retinoblastoma. Ultrasonography is helpful in differentiating the two entities, in that in retinoblastoma a distinct tumor can be detected beneath the retinal detachment, whereas no distinct mass is seen in Coats’ disease (Figure 11.5).
Medulloepithelioma
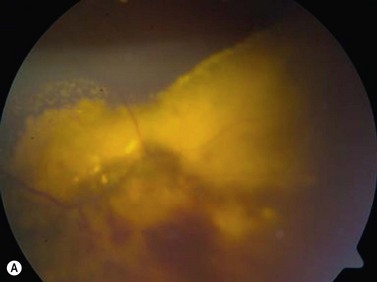
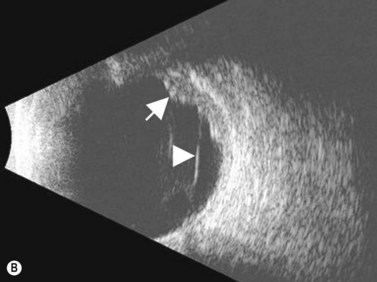
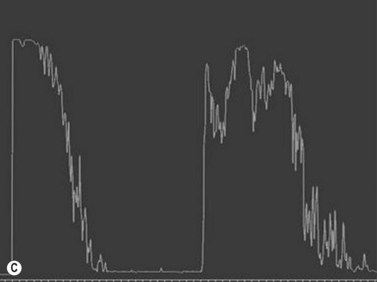
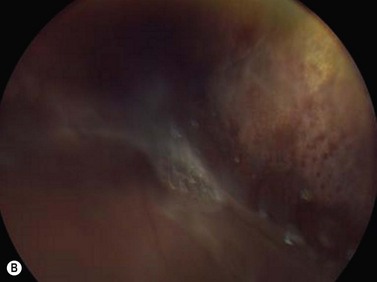
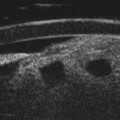
Medulloepithelioma is a congenital neuroepithelial tumor that typically manifests during the first decade of life. It most commonly arises from the ciliary body, however involvement of the iris and optic nerve has also been reported.5–10 On ophthalmic examination, the tumor appears as a lightly pigmented or amelanotic cystic mass. Large cysts may break off from the main tumor and float freely in the anterior chamber or vitreous cavity. Because of their appearance and because medulloepithelioma may present with leukocoria, these tumors are an important consideration in the differential diagnosis of retinoblastoma. A-scan of medulloepithelioma shows mainly high internal reflectivity with a medium spike corresponding to cystic regions of the tumor. On B-scan, medulloepitheliomas typically appear as a dome-shaped, highly reflective mass with irregular internal structures. Cystic spaces can be demonstrated in some lesions (Figure 11.6).
Benign uveal tumors
Circumscribed and diffuse choroidal hemangioma
Clinical features, symptoms, and signs
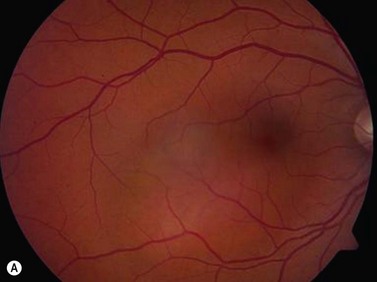
On ophthalmoscopic examination, circumscribed choroidal hemangiomas appear as an orange choroidal mass with indistinct margins that blend with the surrounding choroid. They are frequently located in the macular region of the posterior pole, and are not usually thicker than 6 mm.11 Although these tumors are highly vascular, dilated and tortuous feeder vessels are not typically observed. Surrounding subretinal fluid leading to exudative retinal detachment with macular involvement is common in symptomatic cases. Retinal hard exudates are minimal or absent. Diffuse choroidal hemangiomas appear as orange, diffuse choroidal thickening that has been likened to a “tomato-catsup fundus.” Focal regions of excessively thickened choroid within the diffuse hemangioma may simulate circumscribed choroidal hemangioma. As with circumscribed hemangiomas, there may be associated exudative retinal detachment that often does not become manifest until adolescence.
Diagnostic evaluation
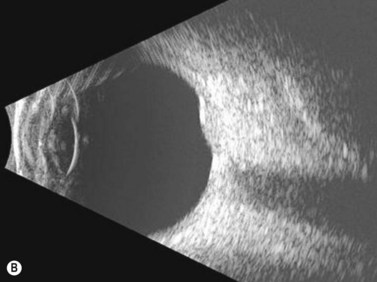
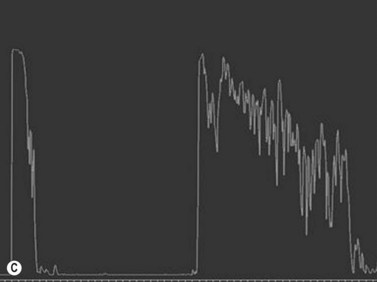
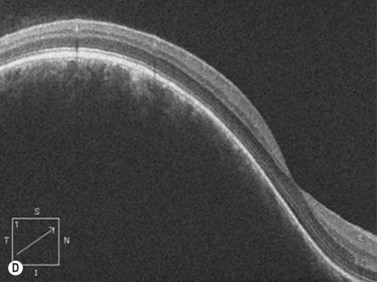
On A-scan, circumscribed choroidal hemangiomas demonstrate high internal reflectivity with negligible attenuation. This differs from other tumors in the differential diagnosis including malignant melanoma which classically demonstrates low to medium reflectivity on A-scan. On B-scan, circumscribed choroidal hemangioma appears as a dome-shaped choroidal mass with smooth contours. They are hyperechoic with regular internal structure and little internal blood flow. Serous retinal detachment at the tumor margins and calcification on the tumor surface may be present.11 Angiographic studies such as fluorescein and indocyanine green (ICG) can also be diagnostic. Fluorescein angiography demonstrates a hyperfluorescent mass with a fine lacy vascular network of intrinsic vessels in the early phases followed by increasing hyperfluorescence throughout the angiogram with variable leakage in late views.12 With ICG angiography, a rapid increase in hyperfluorescence is seen early on followed by a “washout” effect in the late phase.13 OCT can also be helpful in evaluating secondary changes in the overlying retina such as shallow subretinal fluid or cystoid macular edema (Figure 11.7).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


