HISTORICAL BACKGROUND
Paul Wilhelm Heinrich Langerhans (1847-1888) was born in Berlin. His father and his two brothers were physicians. While still an undergraduate, Langerhans used Cohnheim’s gold chloride staining to identify a novel nonpigmentary epidermal dendritic cell. When he was 21 years old, he described this finding in an 1868 paper titled “
Uber die Nerven der menschlichen Haut” (on the nerves of the human skin) (
1). Langerhans initially regarded these cells as intraepidermal receptors for extracutaneous signals of the nervous system. He later changed his mind and in 1882 wrote that “my cells are in no way essential for nerve endings” (
2). Langerhans studied medicine with Heckel and Virchow. He defended his thesis, “
Contributions to the Microscopic Anatomy of the Pancreas.” In 1869, he identified a cell in the human pancreas, now called Langerhans islets. After his thesis defense, Langerhans demonstrated that cinnabar was taken up by white blood cells but not red blood cells. This opened the door for Aschoff’s concept of the reticuloendothelial system.
In 1874, Langerhans became Professor Extraordinarius in Freiburg. Unfortunately, 1 week later he was diagnosed with renal tuberculosis (TB). He was released from his duties at the university and he left for Madeira. He continued to do research on the flora and fauna of the Atlantic Islands and Madeira. His contributions were of such high quality that in 1909 a polychaete worm was named after him. Langerhans practiced medicine whenever his health allowed. The majority of his patients were German and British citizens who lived on the island for health reasons, usually TB. Langerhans died of progressive renal degeneration caused by TB in 1888. He published two papers on TB, undoubtedly influenced by his own condition and by the fact that his mother had died of TB and his half brother had also contracted the disease (
3,
4,
5).
In 1893, Dr. Alfred Hand Jr, a 25-year-old resident at the Children’s Hospital of Philadelphia, reported the case of a 3-year-old boy with “a history of great thirst and polyuria… undersized and puny.”
At autopsy, a yellow spot about the size of a five-cent piece was noticed near the right parietal eminence. When the skull-cap was removed, this spot was seen on the inner side as well, and the entire thickness of the bone there was soft and movable…. The lymphatic glands… all through the body were greatly enlarged…. The liver and spleen were enlarged and firm, and the former had minute gray nodules in its substance…. Microscopial sections showed nodular masses of small, round-celled infiltration in the liver, spleen, kidneys (
5).
Hand suspected that he was dealing with a case of TB. In 1921, he noted the similarity of his original case to ones subsequently reported by Schuller, Christian, and Kay. Eventually, the term
Hand-Schuller-Christian disease or
triad was used to describe a disease occurring in children more than 2 years old characterized by exophthalmos, lesions in the bones of the skull, and diabetes insipidus (DI). The full triad was rarely seen, and the prognosis was good (
6,
7).
In 1924 and 1933, Letterer and Siwe described what they perceived to be an entity distinct from Hand-Schuller-Christian disease.
Letterer-Siwe disease generally occurred in children less than 2 years old. The diagnostic criteria included splenomegaly, hepatomegaly, lymphadenopathy, anemia, and a hemorrhagic diathesis. The prognosis was poor (
5,
6,
7).
In 1940, Otani and Ehrlich (
8) described a granuloma of bone simulating a primary neoplasm. Eventually, the solitary eosinophilic granuloma was described as occurring in children more than 2 years old, characterized by a solitary, usually bony site of involvement, and an excellent prognosis (
6,
7).
In 1953, Lichtenstein argued that eosinophilic granuloma of bone, Letterer-Siwe disease, and Hand-Schuller-Christian disease were related manifestations of a single nosologic entity (
5,
7). He used the name
histiocytosis X to refer to a spectrum of diseases of the mononuclear phagocyte (histiocyte). The
X referred to the unknown etiology and pathogenesis of the disease or diseases. The diseases formerly grouped under the heading “Histiocytosis
X” are now called
Langerhans cell histiocytosis (LCH).
DEFINITION, PATHOGENESIS, AND PATHOLOGY
LCH is an accumulation or proliferation of a clonal population of cells bearing the phenotype of a Langerhans cell that has been arrested in early stage of activation and is functionally deficient. Langerhans cells along with lymphocytes, eosinophils, and normal histocytes form infiltrates typical for the disease, which may be found to a varying extent in multiple or single organ(s). LCH can affect the skin, bone, lymph nodes, ear, gums, lungs, gastrointestinal tract including the liver, and the central nervous system (CNS) by causing DI.
The Langerhans cells are a family of related cells characterized by their dendritic morphology and multiple thinmembrane projections. Dendritic cells constitute 0.2% of white blood cells in the blood and are present in even smaller proportions in tissues such as the skin. Because of their rarity, their true function eluded scientists for nearly a century after
Langerhans first identified them in 1868. In 1973, Ralph M. Steinman of Rockefeller University rediscovered the cells in mouse spleens and recognized that they are part of the immune system (
9). The cells were unusually potent in stimulating immunity in experimental animals. He renamed the cells
dendritic because of their spiky arms, or dendrites. The subset of dendritic cells that occur in the epidermis of the skin are commonly still called Langerhans cells. In normal anatomy, Langerhans cells are found in the epidermis and skin appendages, in squamous mucosal epithelium such as the buccal mucosa, vagina, cervix, and esophagus, and in the spleen and lymphatic system. Dendritic cells attack invading bacteria, digest them, and display their antigens on the surface. Antigen-bearing dendritic cells travel to lymph nodes or the spleen, where they interact with other cells of the immune system, including B cells, which make antibodies, and killer T cells, which attract microbes and ingest them.
LCH cells cause tissue damage by infiltration and excessive production of cytokines and prostaglandins. Cells produce interleukin-1 (IL-1) and prostaglandin E2, which can cause bone resorption through osteoclast activation. IL-1 may cause release of IL-2 and gamma interferon from helper-inducer T lymphocytes, leading to the stimulation of other lymphocytes and histiocytes. Tissue injury results from the local immune response as the collection of immune cells impairs normal tissue structure and function (
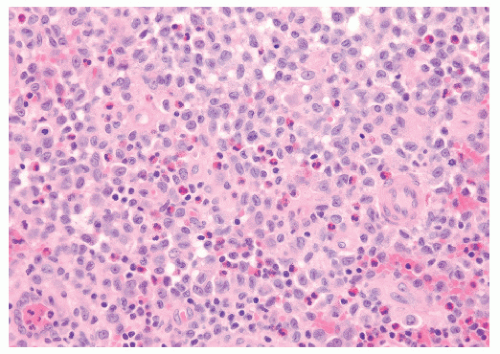
10). On microscopic evaluation, the lesions of LCH are found to be consistent with Hand’s original description: a pleomorphic infiltrate of lymphocytes, eosinophils, polymorphonuclear leukocytes, and Langerhans cells (
Fig. 17.1). A more definitive diagnosis is made when electron microscopy shows Birbeck granules or when light microscopy shows that the Langerhans cells are ATPase positive; stain for S-100 protein, CD11, and CD14, anti-CD1 marker (also called the T6 surface marker), and α-D-mannosidase; and bind peanut lectin (
11,
12) (
Table 17.1). Birbeck granules are membranous cytoplasmic structures, of unknown function, 200-400 mm wide and shaped like tennis rackets (
11,
13,
14) (
Fig. 17.2).
Lesional cells of LCH may represent Langerhans cells arrested in early stage of activation. Immunohistochemical expression of fascin, a protein that is a marker for dendritic cells, is observed in many cases of LCH. This immunoreactivity for fascin supports LCH’s pathogenesis from cells of the dendritic system (
12).
Until recently, the disease generally was not thought to be monoclonal in origin. It was argued that the individual cells do not show atypia and that the disease lacked the usual histologic criteria for malignancy. Therefore, LCH was generally considered to fall within the realm of reactive immunologic disorders or perhaps to be of infectious origin. However, viral and immune causation theories generally have lacked supporting evidence. In 1994, Willman et al. (
15) provided evidence that LCH is a clonal proliferative disorder. These investigators studied 10 lesions from patients with LCH and found that they all contained clonal populations of cells. The proportion of clonal cells corresponded to the proportion of lesional Langerhans-like cells, whether from solitary lesions or extensive multisystem disease (
16). Yu et al. (
17) reported similar findings by flow-sorting CD1a-positive cells from three patients. The hypothesis that LCH arises from somatic mutation of DNA in a normal Langerhans or precursor cell must be considered (
16). The evidence of a genetic mutation or mutations leading to LCH is circumstantial. On average, patients with multiorgan LCH are younger than those with single-bone disease. In the rare instances of familial LCH, all the affected members have multiorgan disease. These two observations,
reminiscent of the pattern of heritable versus nonheritable retinoblastoma, suggested to Egeler the possibility that LCH’s behavior was explicable by Knudson’s two-hit hypothesis and a tumor suppressor gene (
18). (The two-hit hypothesis is discussed in detail in
Chapter 5.)
The distinction between calling LCH malignant and considering it a reactive immunologic disorder is important. Classification has significant consequences for how the clinician thinks about and manages the disease. If one believes that LCH is primarily a malignancy, then radiation therapy (RT), cytotoxic chemotherapy, and even bone marrow ablation and transplantation seem to be acceptable primary therapies. However, if one thinks about LCH primarily as a reactive disorder, then at least initially one would be likely to use more conservative and less toxic therapies, including expectant observation. For the clinician, the truth lies between these two classifications. Most cases of LCH are not lifethreatening and can be managed with minimal therapy. There are a few instances of the disease running a fulminant and fatal course, a situation that calls for an aggressive response.
CLINICAL PRESENTATION, DIAGNOSTIC EVALUATION, AND STAGING
The annual incidence of LCH is approximately 0.5 to 2.0 cases per 100,000 children per year. There is a male predominance, with 56-66% of patients male, and 50% of cases diagnosed between the ages of 1 and 15 years (
12,
13,
19,
20,
21).
The form of clinical presentation is related to the child’s age (19-33) (
Tables 17.2 and
17.3). In children less than 2 years old, there may be a widespread seborrheic rash (
Fig. 17.3). The rash is often most pronounced on the scalp and in the groin. Petechial hemorrhages in the involved skin are characteristic. Erosive intertrigo in the groin, axilla, and perianal region is a common feature. Ulceration and secondary skin infection may occur. Involvement of the mastoid and middle ear may present as a chronic draining otitis. The infant may be irritable, with a diminished appetite and failure to thrive. Palpable lymphadenopathy may be seen secondary to LCH infiltration. Liver involvement is common in disseminated LCH. Hepatomegaly, elevation of liver enzymes, increased
conjugated bilirubin, ascites, edema, and failure to thrive may be attributable to liver disease or involvement of the gastrointestinal tract. Diarrhea may be the result of abnormal bile acid metabolism or malabsorption (
14). Splenomegaly may be accompanied by anemia, leukopenia, or thrombocytopenia (
39). In children more than 2 years old, the most common presenting symptoms are related to bone involvement (
Table 17.3). There is localized pain, with or without an associated soft tissue mass. Involvement of the orbital bones may cause exophthalmos. LCH may produce premature eruption of the teeth or tooth loss because of gum and mandibular disease (
39). Back pain and loss of vertebral height may be seen. Spinal cord compression is rare.
DI is the most common complication of CNS involvement. The incidence varies, in different reports, from 11% to 50%. Magnetic resonance imaging (MRI) may show lesions in the posterior pituitary or the pituitary stalk (
40). The mechanism of injury is thought to be either infiltration of the meninges adjacent to the posterior hypothalamic-pituitary axis or direct involvement of the brain. DI often is associated with skull lesions, which may be seen before the DI (
23,
38,
41,
42,
43,
44). This may be important because if the index of suspicion for DI is high, and it is diagnosed early, then a few authors believe prompt irradiation may reverse or ameliorate the DI. Lung and oral mucous membrane involvement with LCH often occurs in patients in whom DI develops (
41). In a Dutch-German-Austrian study, 3 of the 93 patients (3%) with primary localized disease and 16 of the 106 patients (15%) with dissemination of LCH at diagnosis had DI (
41). In a series of patients from London’s Hospital for Sick Children, DI was more common among children with multisystem disease (12 of 32) than among those with disease apparently confined to bone (3 of 20). Fourteen of the 15 children with DI had bone disease involving the skull. The cumulative risk of developing DI during the first 4 years after presentation with LCH was 42% (
42). In a series from San Francisco, 25% of patients developed DI (
25).
Pulmonary LCH has been reported in infants and older adults. However, it affects primarily adults in their twenties and thirties. Chest CT demonstrates multiple nodules with a predominance in the upper lobes. If bronchioalveolar lavage yields more than 5% LCH cells, then the procedure is diagnostic. The cells are CD1a positive. In adults, the natural history of LCH is variable. It is often associated with cigarette smoking, and patients who continue to smoke progress to end-stage fibrotic disease or develop extrapulmonary complications. Smoking cessation is the most effective therapy (
45).
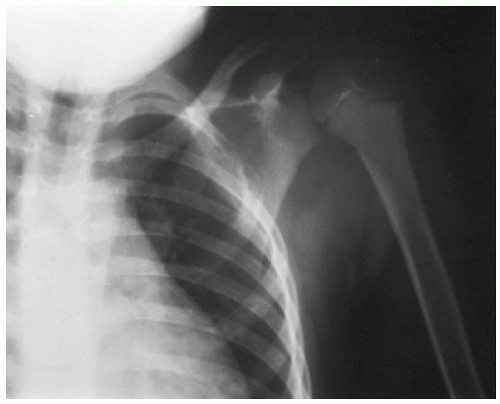
Skull lesions often are the first sign of LCH. Often very slow growing, they may be single or multiple and can occur without other bony involvement. Although they have a predilection for the temple area, they can be found anywhere on the skull. On plain radiographs they will appear as punched-out lesions. With time they can develop a sclerotic border (
46). Calvarial lesions can have epidural extension. They may extend beneath the dura into the brain parenchyma.
A diagnostic radiograph skeletal survey should be performed to assess the extent of bony involvement (
Fig. 17.4). In LCH, conventional skeletal surveys are complementary to isotopic bone scans (
46). Both are often used (
47). In skeletal radiographs, LCH produces a focal area of rarefaction. The lucent area begins with the medullary cavity and extends to
involve the inner table of the cortical bone. MRI may show that the area of bone abnormality is larger than suspected from other studies. Preliminary data suggest that PET scanning may identify active lesions and differentiate them from healed lesions after treatment (
48).
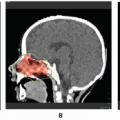
In disseminated LCH, cerebral involvement may occur (
49). There can be meningeal involvement with formation of large plaques of subdural tumor and/or intraparenchymal lesions. The most common sites for intraparenchymal disease are the hypothalamus and cerebellum. Less common locations are the frontal and temporal lobes (
50). Occasionally, LCH can first present to medical attention as an isolated, unifocal lesion of the CNS. The hypothalamus is the most common site for this rare situation. In such cases, the diagnosis is made after surgical exploration and biopsy (
49,
50,
51).
LCH has a variable prognosis, and a system of grouping patients by predicted outcome is a valuable guide for the clinician (
52,
53,
54). Patients with localized disease (skin, bone, or lymph node) have a good prognosis and often require minimal or no treatment. In contrast, multiple organ involvement, which is particularly frequent in children <2 years of age, has the risk of a poor prognosis (
Table 17.4). Lahey and coworkers (
29,
30,
55) found a striking difference in survival between patients with and without organ dysfunction. Of the 50 patients without organ dysfunction, 33 (66%) responded to chemotherapy and 2 (4%) died. In 33 patients with dysfunction of one or more of the three organ systems, only 11 (33%) responded to chemotherapy, and 22 (67%) died.
The stage distribution of LCH at presentation is almost certainly influenced by referral patterns. A children’s hospital is likely to see a higher proportion of advanced disease than a community hospital. In the Children’s Hospital of Philadelphia series, 33 of the 64 patients (52%) had localized disease at presentation, 22 (34%) had multifocal disease without organ dysfunction, and 9 (14%) had multifocal disease with evidence of organ dysfunction (
21). In a series by McLelland et al. (
22) of 58 children, 14 (24%) had single-system disease, 22 (38%) had multisystem disease without organ dysfunction, and 22 (38%) had multisystem disease with organ dysfunction (
22).