Fig. 20.1
Section of the cingulate cortex of an Alzheimer patient (F, 84 years) stained for amyloid plaques with the Gallyas modification of the Bielschowsky silver method. Upper panel shows numerous plaques in all cortical layers except layer 1. Lower panel gives a higher magnification of a single plaque with the fuzzy appearance of dense amyloid fibrils (Luiten and Majtenyi, unpublished results)
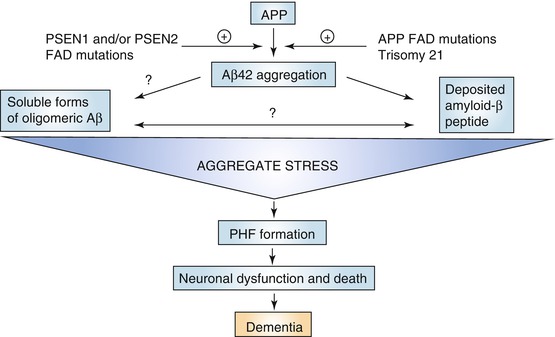
Fig. 20.2
Diagram of the amyloid cascade hypothesis of Alzheimer’s disease. Starting point of this theory is the formation of Aβ peptide as a splicing product of the amyloid precursor protein (APP) by the activity of β- and γ-secretases. The activity of these enzymes can be affected by mutations in the presenilin genes (PSEN1 and PSEN2) which is considered as a major cause of genetic forms of AD, next to mutations in the APP gene on chromosome 21 and trisomy of chromosome 21 (Down syndrome). When not timely removed or cleared, Aβ aggregates initially to neurotoxic oligomers (“aggregate stress”) eventually leading to large-scale neuronal cell death as a result of excessive pathological tau phosphorylation. These phosphorylated tau complexes take the shape of paired helical filaments (PHF) which accumulate in the cell bodies of affected neurons. During that process neurons lose their function and eventually die in great numbers. At that stage dementia symptoms become manifest (Reproduced with permission from Karran et al. (2011))
Taking Aβ as starting point of therapeutic strategy, it is not surprising that great efforts were made to follow the amyloid deposition process in the living brain (Villemagne et al. 2008; Quigley et al. 2011).
Alzheimer himself already described the presence of amyloid as numerous, fuzzy depositions of a peculiar compound (the “eigenartige Stoff” he mentioned), which now has been characterized as consisting of the protein fragment Aβ. We know that Aβ is the splicing product of a much larger amyloid precursor protein (APP) by the sequential enzymatic activity of two enzyme complexes, beta-secretase and gamma-secretase. This results in the production of the amyloid peptides, mainly consisting of 40- and 42-amino acid residues (De Strooper et al. 1998; Vassar et al. 1999). In particular the Aβ 42 fragment has the tendency to cluster into aggregates, initially as soluble oligomers (Masman et al. 2009; Ono et al. 2009), but this process continues and ends up with the formation of larger fibrils. The fibrils, during a process that can last several years, collect in the shape of the well-known fuzzy plaques described by Alzheimer. These amyloid plaques can be visualized in postmortem brain sections of AD patients by means of classical silver staining methods like the Bielschowsky techniques (Fig. 20.1). The amyloid plaques not only contain amyloid fibrils but also consist of degenerating neuritic profiles including pathological tau accumulation, activated microglia, and other neural components associated with neurodegenerative mechanisms (Jellinger 1998; LaFerla 2010). For that reason the amyloid plaque for a long time was considered as the primary locus of a neurodegenerative and neurotoxic cascade, but this idea has to be abandoned. There is now strong evidence that not the insoluble plaque but the initial soluble Aβ oligomers appear to be the neurotoxic form of amyloid (Shankar et al. 2007; Walsh and Selkoe 2007).
Of course we can visualize amyloid and Aβ plaques in the postmortem brain of the AD patient (Fig. 20.1), but can we also demonstrate the amyloid formation in the living brain, preferably in an early stage of the disease? Early amyloid detection has always been an issue of high priority since early detection would enable studying anti-amyloid treatment effects prior to the dramatic destruction of brain tissue in the advanced stages of the disease (Nordberg 2010; Miller 2012). To that aim, a number of compounds have been developed that allow detection of amyloid in the living brain by means of PET imaging (Fig. 20.3). One of the first PET tracers for Aβ detection was the “Pittsburg compound B” labeled with 11C (11C-PIB) (Rabinovici et al. 2007; Nordberg et al. 2010; Nordberg 2011), with a chemical structure derived from thioflavin-T that is commonly used for the detection of aggregated amyloid in the postmortem brain. 11C-PIB proved to be a tracer by which amyloid could be detected in vivo in patients diagnosed with AD (Resnick et al. 2012) (Fig. 20.3). However, different results were obtained when applied to patients with mild cognitive impairment that is often considered as an early clinical stage of ongoing AD pathogenesis (Okello et al. 2009). Since the discovery of PIB, also several other PET tracers for Aβ have been generated such as florbetaben (18F). Florbetaben has a high binding sensitivity to Aβ, but not to aggregated alpha-synuclein or phosphorylated tau and consequently allows a more reliable and differential diagnosis (Barthel and Sabri 2011). A major advantage of ligands like florbetaben labeled with 18F is the much longer half-life of the radioactive label, which enables PET tracing without the immediate presence of costly cyclotron facilities (Frisoni 2011). It is obvious that the clinical application of PET tracers serves diagnostic purposes and disease development but also allows longitudinal study of therapeutic treatment strategies.
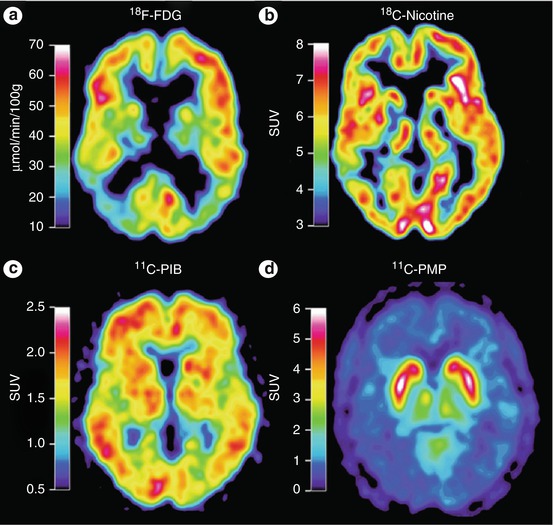
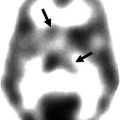
Fig. 20.3
PET images of an AD patient subjected to tracer study using 18F-FDG for glucose metabolism, 11C-nicotine for nicotinic receptor binding, 11C-PIB for amyloid imaging, and 11C-PMP for revealing acetylcholinesterase activity. The images show a low level of metabolic activity (FDG), reduced binding of nicotinic receptors, and high binding levels of the amyloid tracer PIB, in particular in the left temporal lobe. PMP images are low in the entire cortical mantle indicative of large-scale cholinergic denervation. Abbreviations: FDG 2-fluoro-2-deoxyglucose, PIB Pittsburg compound B, PMP N-methylpiperidin-4-yl propionate (Reproduced with permission from Nordberg et al. (2010))
Clinical applications are only possible after extensive preclinical and experimental testing of drugs in innovative development programs. In that respect the demonstration of 11C-PIB binding in amyloid-rich regions of transgenic mouse models of AD proved to be of considerable scientific value (Maeda et al. 2011; Manook et al. 2012). In mice overexpressing a mutated human APP gene, Maeda and colleagues showed high affinity binding of 11C-PIB. Interestingly, the degree of binding with PET directly correlated with plaque density as demonstrated with 11C-PIB autoradiography, which progressively increased with the age of the animals (Manook et al. 2012). Moreover the PIB binding was shown to be Aβ-subtype specific. By applying this PET tracer, a reduction of amyloid plaque load could be demonstrated after the treatment of the transgenic mice with an antibody against Aβ (Maeda et al. 2007). Apart from diagnostic purposes, the application of PET ligands for Aβ detection can be of great value for evaluating potential Aβ-modifying or Aβ-decreasing drugs and other therapeutic approaches. An essential question, however, remains as to the state in which Aβ can be visualized by the currently available PET tracers (Ikonomovic et al. 2012).
20.4 Amyloid Neurotoxicity
Recent insight in amyloid biology strongly indicates that the very early stages of amyloid production and subsequent aggregation to soluble oligomers may represent the neurotoxic phase of the pathogenesis in AD (Walsh and Selkoe 2007). How precisely small Aβ aggregates and by which mechanisms oligomers exert their toxic effects on brain tissue is still far from clear. However, several lines of evidence point to an interaction of Aβ with glutamatergic synaptic signaling, which is notably prominent in the cognitive regions of the forebrain (Shankar et al. 2007). Our own studies in that respect demonstrated that exposure of cultured astroglia to amyloid peptides leads to a highly significant suppression of the uptake of glutamate by astrocytes possibly by blockade of the glutamate transporter (Harkany et al. 2000c). Such an effect of amyloid on astrocytes would create a glutamate concentration increase in the amyloid microenvironment. This was confirmed by our microdialysis experiments in which we infused amyloid oligomers into the brain parenchyma with simultaneous sampling of extracellular fluid via the same microdialysis probe. HPLC analysis of these samples clearly showed a steep rise in glutamate concentrations as a direct consequence of the amyloid injection (Harkany et al. 2000c). The neurotoxic consequences of amyloid-induced glutamate concentrations can partly be associated with an excitotoxic cascade, which appears to be a common feature of many neurodegenerative diseases (Lipton and Rosenberg 1994). Uninhibited overstimulation of the glutamatergic synapse notably by continuous activation of the NMDA receptor will result in intracellular rise of the calcium concentration and loss of calcium homeostasis followed by a complex excitotoxic sequence of injurious events (Harkany et al. 2000b). Further evidence for such a neurotoxic process triggered by Aβ was supplied by calcium accumulation in amyloid injection areas as demonstrated by 14C-calcium autoradiography. Also several other studies lend support to the injurious impact of amyloid on the NMDA receptor (Mattson 2004; You et al. 2012) and its consequences for activating calcium-related neurotoxic pathways (Granic et al. 2010a; Zempel et al. 2010).
It appears that the sequence of amyloid deposition, glutamate overexposure, accumulation of intracellular calcium, and calcium-induced neurotoxicity brings the amyloid environment in a neurotoxic vicious cycle. This can be concluded from the fact that exposure of the brain tissue to glutamate and NMDA triggers the expression of APP which most likely will enhance Aβ production (Harkany et al. 2002). The great challenge for molecular imaging would be to detect and visualize the amyloid aggregation processes in very early stages of the disease since they represent the prodromal and presymptomatic conditions of AD (Miller 2012).
20.5 Combating Amyloid Plaque Load: The Way to Go?
The continuing aggregation process of Aβ over many years eventually will generate deposition of insoluble amyloid fibrils in the form of the classical amyloid plaque as observed already by Alzheimer. In that sense, the amyloid plaque should probably be considered as an inert rest product of a long-term aggregation process without a serious role in a neurotoxic cascade of events. For a long time, one of the major strategies of drug development has been and still is the development of therapeutic approaches aimed at reducing the amyloid burden and plaque load. There are several approaches towards such aims including inhibiting secretases to reduce the production rate of Aβ (Siemers et al. 2006) or strategies aimed at interference with the aggregation process (Gandy et al. 2003; Chen et al. 2009; Karran et al. 2011). In the latter way of thinking, several compounds were designed, derived from the native amyloid amino acid sequences designated as beta-sheet breakers (BSB), that would be able to cleave the bonds between the aggregated Aβ peptides. In particular, the Soto group developed families of small peptides (tetra- or pentapeptides) with high specific binding to the native Aβ. These peptides were designed to prevent the amyloid-amyloid beta-sheet formation or to break down the aggregated amyloid molecules (Soto and Estrada 2005; Chen et al. 2009). Following this strategy we carried out a series of experiments in which we tested the impact of exposure of brain slices endowed with amyloid plaques with selected BSB pentapeptides with the sequences LPYFD and RVVIA (Granic et al. 2010b). Brain slices were obtained from the temporal lobe from postmortem AD and from transgenic APPxPS1 mutant mice, prepared for organotypic slice culturing, and stained for aggregated Aβ with thioflavin-S. By confocal microscopy slice regions with plaques were selected and incubated with BSBs for 20 h during which every hour the integral plaque density was measured and calculated. Plaques exposed this way to BSBs, both in postmortem human AD (Morley and Farr 2012) and transgenic mice, showed a linear reduction of the three-dimensional thioflavin-S signal of up to 40 % as a result of the BSB activity.
Within the limitations of this type of experiments, these data provide proof of principle that small peptides derived from the Aβ sequences are able to reduce the plaque size. The underlying mechanism could be the dissolution of the aggregated fibrillary amyloid or breaking the intermolecular bonds between the amyloid entities. However, it remained unclear how the potential splicing products within this experimental setup would influence the surrounding neural tissue, but our expectations were not too high. The dense amyloid fibrils in the plaques should probably be considered as an insoluble and chemically inert rest product of a long-term polymerization process. As such the amyloid plaques may not present a threat to adjacent neuronal structures. It may even well be that “dissolving” amyloid polymers might generate small molecular entities with neurotoxic properties, although we have no information that supports such a pathway. However, very recent data on the outcome of clinical trials with the anti-amyloid antibody “bapineuzumab” (Rinne et al. 2010) point to a similar conclusion. In this phase 2 clinical investigation, moderate AD patients were treated with this antibody up to 78 weeks during which PET scans were made with the amyloid tracer 11C-PIB at various timepoints. Major findings were that PIB retention increased during the trial period in the placebo group while the antibody treated cohort showed a reduction of 11C-PIB binding resulting in a significant reduction in the PIB retention profiles. This reduction was interpreted as a diminished fibrillar Aβ load in the cortical regions analyzed. However, due to a lack of behavioral effects and improved dementia scores, the clinical trial with this antibody was halted in this group of mild to moderate AD patients (www.alzforum.org Aug 13, 2012) (Miller 2012). From this study it may be concluded that reduction of plaque load will not necessarily mean an improvement in cognitive performances. The conclusion seems also justified that treatment of patients with an ongoing and progressive pathological process in which dementia has become manifest is not to be expected to yield significant behavioral relief. Also once more these findings warrant very early and prodromal diagnosis and design of therapies aimed at preventing the very early formation of probably soluble amyloid species.
20.6 Novel Anti-amyloid Approaches
In spite of disappointing data with approaches aimed at reducing the amyloid plaque load as reported above, the fact remains that amyloid peptides in oligomeric conformations are toxic to nerve cells (Shankar et al. 2007). However, the mechanisms through which Aβ induces cell death or cell damage are not well understood. The signaling proteins associated with Aβ toxicity, including receptor proteins, are only partly known (Huang and Mucke 2012; You et al. 2012). Neuronal cell death induced by Aβ oligomers can easily be demonstrated by applying Aβ to cortical neurons in culture thereby showing dose-dependent degrees of neuronal cell death. These cell culturing techniques are attractive tools for fast testing of potentially neuroprotective drugs in AD. An example of such developments comes from our own studies on the molecular dynamics (MD) of Aβ in dimer and pentamer conformations (Masman et al. 2009). With these MD simulations, it was possible to observe the conformational behavior of the amyloid aggregates in solution, thus detecting their strongest and weakest intermolecular interactions. Therefore, with this valuable information we designed a novel generation of compounds, which could interfere with the aggregated Aβ at the beta-sheet domains of the molecule. According to these MD calculations, the most energetically favorable interactions were selected at the levels of the amino acid residues glutamate 22 and aspartate 23. These strong interactions of putative drugs with the amyloid aggregates might weaken the amyloid-amyloid interactions, thus destabilizing (or impeding) further growth of the amyloid fibrils. Based on these findings, we generated a family of pentapeptides derived from the Aβ sequences, but possessing modified amino acid residues in order to increase their lipophilicity) (Fig. 20.4) (unpublished data).
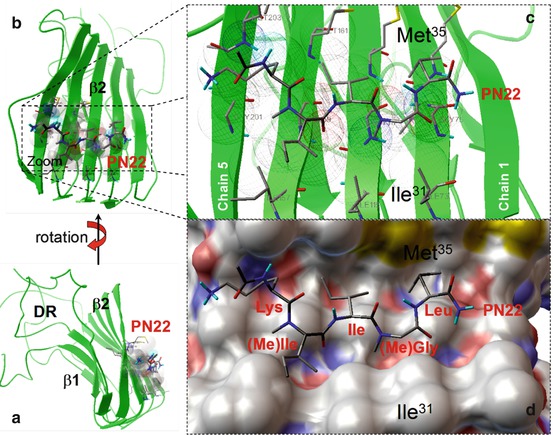
Fig. 20.4
(a) Stereoviews of an Aβ oligomer of five amyloid molecules (pentamer) based on molecular dynamic calculations. The oligomers take a well-organized structure consisting of a β1 and a β2 strands connected via a hairpin loop. In the β-sheets, the amyloids run parallel and display strong molecular interaction with low energy levels. The remainders of the amyloid molecules show few interactions and are designated as the disordered regions (DR). (b) Shows a lateral view of the oligomer with the site of strongest intermolecular bonding and the location of the “anti-amyloid pentapeptide” PN22. The PN22–β2 complex is enlarged in panels (c, d) to show more detail of the binding pattern. Panel (c) shows the ligand PN22–beta 2 target contacts which are depicted as wireframe spheres. Panel (d) shows a stereoview of PN22 in the channel-like hydrophobic pocket formed, mainly by residues Ile31 and Met35 (Abbreviations: Ile isoleucine, Leu leucine, Lys lysine, (Me)Ile methyl isoleucine, (Me)Gly methyl glycine, the numbers of the amino acids refer to their numerical position in the amyloid molecule)
Pentapeptides with highest potential in the MD simulations were synthesized and tested for their neuroprotective effects against Aβ oligomers both in neuronal cell culture and in vivo in mouse hippocampus. One of these pentapeptides, which we coded PN22 proved to be particularly effective and dose dependently protected against Aβ-induced neuronal cell death up to 90 %. When injected into the mouse dorsal hippocampus, Aβ oligomers caused severe memory loss as measured by fear conditioning, which could be antagonized by applying PN22 to the injected amyloid oligomers. Although these data raise many questions as to the nature of the neuroprotective action of PN22, these preliminary data may open new avenues towards novel anti-amyloid drug development. These findings also indicate the need of in vivo imaging tools and tracers that could visualize the very early stages of amyloid production prior to overt damage to nervous tissue. It is worth mentioning at this point that these types of compounds, after appropriate chemical transformations, are excellent candidates to become PET tracers due to their high affinity for amyloid aggregates and also because their structure possess several opportunities for radioactive labeling. There are four positron-emitting radioisotopes that are used more than any other in PET imaging, namely, fluor-18, carbon-11, nitrogen-13, and oxygen-15. All these elements are normally present in peptide structures giving virtually an infinite number of possibilities to include them in the chemical structure, such on the condition that these substitutions do not significantly alter the chemical properties of the native compound. Fluorine-18, for example, can be easily included as a substitution of a nonreactive hydroxyl group (Schlyer 2004).
20.7 Aβ Production and Clearance Mechanisms of Aβ
A question that has been posed from the beginning of amyloid research is whether the Aβ fragments of the amyloid precursor is a pathological aberration or a molecular product with a neurobiological or neurophysiological function (Luo et al. 1995; Morley and Farr 2012). There are now several arguments to support the view on Aβ as a normal biological phenomenon, which may derange due to a variety of environmental or genetic factors. Why is amyloid increasing in later phases of life (Huang et al. 2012)? Aβ, like any protein in the brain, after its production survives for a limited amount of time and is subsequently inactivated and removed by a number of clearance mechanisms. In general terms, when the production of Aβ increases or when clearance decreases, the balance is disturbed and the protein can accumulate in the brain parenchyma (Karran et al. 2011). For a long time, the general notion was that breakdown of the amyloid precursor protein APP by alpha-secretases resulted in soluble fragments. Problems would start when APP was spliced at different locations of the APP molecule by the activation of beta- and gamma-secretases (De Strooper et al. 1998; Karran et al. 2011). One of the most striking recent findings with respect to amyloid production was reported by the Bateman group in a number of significant papers (Bateman et al. 2006). These authors analyzed the production and clearance of Aβ in AD patients and in age-matched control subjects. This was achieved by systemic infusions of leucine as an integral amino acid component of the Aβ peptide in which the leucine was labeled with 13C6. Production and clearance of labeled Aβ was followed by the analysis of hourly samples from the cerebrospinal fluid (CSF). The analysis was based on immunoprecipitation and mass spectrometry of CSF samples. The most striking outcome of this study was the fact that Aβ production was similar between AD patients and control individuals but that the clearance rates were significantly lower in the AD group compared to control subjects, both for Aβ40 and Aβ42 (Mawuenyega et al. 2010). This difference between production and clearance of Aβ over many years may well account for the accumulation of Aβ and its subsequent plaque formation as a final extracellular deposit phenomenon. A most important conclusion drawn by Mawuenyega et al. in their “Science” paper was that an early detection of a dysbalance between production and clearance would be of great significance for early presymptomatic diagnosis (Mawuenyega et al. 2010).
20.8 Aβ Clearance and Transport Over the Blood–brain Barrier
This important finding by the Bateman group brings us to the dynamics of Aβ clearance. Aβ is cleared by the brain via several mechanisms including enzymatic processing (e.g., by neprilysin, several metalloproteases, insulin-degrading enzyme), intracellular breakdown mechanisms, and transporting Aβ out of the brain (Miners et al. 2011; Sagare et al. 2012). Aβ can be transported out of the brain parenchyma via perivascular pathways into the cerebrospinal environment and via active receptor-mediated transport over the blood–brain barrier. The latter processes appear to be highly complex (Zlokovic 2011). The endothelial lining of the brain vasculature in that respect is of paramount importance. The endothelium is characterized by the specific anatomical structure of tight junctions and the paucity of vacuolar bulk transport options (Farkas and Luiten 2001). Successful transport in nonpathological conditions can only be achieved by the activity of transmembranous carrier proteins which are numerous. Of great importance for amyloid clearance are some of the ATP-binding cassette (ABC) transporters, including P-glycoprotein, breast cancer resistance protein (BCRP), and the lipoprotein receptor-related protein-1 (LRP1). Notably, LRP1 has been identified as a major active efflux route for Aβ through which the peptide can be released into the bloodstream and subsequently be removed by systemic clearance in the liver (Sagare et al. 2012). However, a reverse route mediated by the receptor for advanced glycation endproducts (RAGE) allows transport of Aβ from the bloodstream into the brain parenchyma (Deane et al. 2004).
Furthermore, in recent years the relevance of the ABC transporter Pgp turned out to be associated with neurodegenerative processes and has become an important research topic for in vivo study. This transporter can well be labeled by PET techniques with the tracer 11C-verapamil both in human and in experimental animal brain (Maeda et al. 2007). Pgp can be of importance to AD since this amply expressed that transporter can pump Aβ out of the endothelium into the bloodstream and poses a barrier function of blood-borne compounds like Aβ inhibiting its entry into the brain (Lam et al. 2001). Several studies have reported a reduction of the activity of the Pgp transport system in ageing and in AD, which was demonstrated by an increased verapamil binding corresponding to a lower Pgp function (Fig. 20.5) (Bartels et al. 2009). Recent neuropathological studies moreover revealed a significantly lower expression of Pgp in the microvasculature in AD (Wijesuriya et al. 2010). Experimental investigations of the Pgp transporter with regard to amyloid balance were performed by Cirrito and coworkers (Cirrito et al. 2005). These authors measured the removal out of the brain of radioactive-labeled Aβ in intact wild-type control mice and in mice with double knock out (k.o.) of the MDR1 genes, of which Pgp is the protein product. The Pgp k.o. animals showed a strong reduction of transporting Aβ out of the brain which was calculated as a significantly lower clearance rate of Aβ in the Pgp-k.o. mice. These and other data lend strong support to the notion that Pgp, which is highly expressed in the luminal side of the endothelium in the brain, is of great importance in Aβ clearance mechanisms.
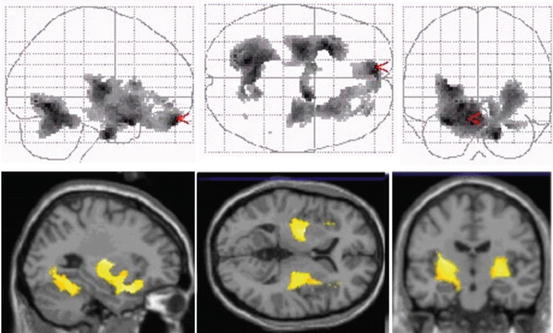
Fig. 20.5
Projected PET images of 11C-verapamil scans in sagittal, horizontal, and coronal planes showing increase of binding in the aged brain (avg. 60 years) compared to young brain (avg. 24 years). The increased binding is interpreted as a reduction in P-gp activity in the aged (Reproduced with permission from Bartels et al. (2009))
20.9 Microvascular Breakdown in Ageing and AD
There is growing awareness that the condition of the cerebral vasculature is a primary factor for brain function in ageing and in neurodegenerative disease. In our lab we carried out an extensive study of the ultrastructure of small brain vessels in a number of mammalian species including human and the effects of ageing and the condition in neurodegenerative disease (Farkas and Luiten 2001). These studies mainly focused on cortical microvessels and capillaries since these vessels, due to their large number and high tissue density, constitute the blood–brain interface and blood–brain barrier. These investigations demonstrated that brain microvessels in all mammalian species including man are subject to an almost linear increase of aberrations within the vascular wall. These aberrations consist of thickening of the basement membrane, swelling of the surrounding astrocytes, increase in endothelial diameter, perivascular deposition of collagen, and a general deformation of the vascular lumen (Grammas et al. 2011). Such anomalies have serious consequences for cerebral flow dynamics, add to the observed reduction of cerebral blood flow, and impair exchange of blood-borne compounds to brain and reversely from brain to general circulation (Farkas and Luiten 2001) (Fig. 20.6).
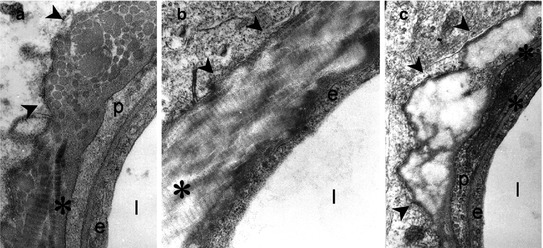
Fig. 20.6
Electron microscopy of microvascular degeneration as occurs in the cingulate cortex observed in aged rat, rhesus monkey, and human, characterized by deposition of collagen in its fibrillary (a, b) or amorphous (c) configuration. This type of microvascular degeneration is more than doubled in Alzheimer’s disease. Collagen depositions are found in the perivascular basement membrane (asterisks) surrounding the endothelial lining of the vessel (e). Abbreviations: p pericyte, l vascular lumen. Arrowheads point to various forms of collagen depositions in the basement membrane layer (E. Farkas and P.G.M. Luiten, 2000. Unpublished)
The importance of the capillary condition was further indicated by experimental animal data in which we quantified the effects of chronic hypoperfusion on microvascular degeneration which correlated well with a cognitive decline as measured in memory tests (Farkas et al. 2007). Next to the fate of microvessels during normal ageing, we found that degenerative changes in brain capillaries in the cingulate cortex of AD patients but also of patients with Parkinson’s disease and Lewy body dementia were more than doubled (Farkas and Luiten 2001). In summary we can conclude that the ultrastructural pathology of microvasculature in neurodegenerative conditions like AD has severe consequences for cerebral blood flow and transport mechanisms over the BBB. There is little doubt that microvascular degeneration in AD is a prominent factor in the early development of the disease and thus a topic that warrants extensive study in patients and animal models for which PET scanning is a tool of choice.
20.10 Microvascular Cholinergic Innervation in the Cortex in Alzheimer’s Disease
In the previous section, we described the breakdown of the microvasculature in the cerebral cortex in animal and man and in AD. At the same time the blood supply to the brain and forebrain in particular is prone to significant reductions both in the ageing individual, but even more so in the cognitively impaired human (Buckner et al. 2000). A diminished flow can mean many things and the consequences are far from clear. It can be argued that unhampered supply of nutrients, notably glucose and oxygen, is of paramount importance for energy generation to the functionally active neuron. Any brain activity readily demands direct production of energy and ATP for maintenance of ionic balances of the nerve cell, signal transduction, and metabolic stability. Even very local activity of nervous tissue should lead to the local increase of blood circulation, and damage to the vascular control mechanisms has dramatic consequences as we know from stroke research. As described above, we know that the vasculature is prone to develop age-related aberrations and that blood flow becomes less with increasing age and in AD. What may be the underlying causes of these phenomena? Experimental and pathological studies reported that next to advanced age and certain genetic dispositions, hypertension, diabetes, food composition, diet and fatty acids, blood-borne amyloid, and hormones, all can play a role in vascular mechanisms that eventually lead to low cerebral circulation.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


