Peripheral nerve enlargement may be seen in multiple conditions including hereditary or inflammatory neuropathies, sporadic or syndromic peripheral nerve sheath tumors, perineurioma, posttraumatic neuroma, and intraneural ganglion. Malignancies such as neurolymphoma, intraneural metastases, or sarcomas may also affect the peripheral nervous system and result in nerve enlargement. The imaging appearance and differentiating factors become especially relevant in the setting of tumor syndromes such as neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. This article reviews the typical magnetic resonance neurography imaging appearances of neurogenic as well as nonneurogenic neoplasms and tumorlike lesions of peripheral nerves, with emphasis on distinguishing factors.
Key points
- •
MR imaging characteristics of many lesions in and around the peripheral nerves are unique and a confident diagnosis of such lesions including lipoma, fibrolipoma, ganglion cyst and perineurioma can be prospectively suggested.
- •
Multiple MR imaging findings can suggest the possibility of a neurogenic tumor but do not reliably distinguish between schwannoma and neurofibroma.
- •
Although there are certain MR features that increase the likelihood of MPNST, biopsy is still necessary to confirm the diagnosis.
- •
Understanding of the clinical features of a broad range of neuropathy conditions and a multidisciplinary approach is essential for proper and timely patient management.
Introduction
Patients with signs and symptoms of focal neuropathy are often referred for nerve imaging, which may consist of regional magnetic resonance (MR) imaging of different areas of the body. The imaging may be designed to provide an overview of tumor burden (whole-body MR [WBMR] imaging) or detailed anatomy of a specific nerve (MR neurography [MRN]). When peripheral nerve enlargement is seen, there is a common differential that includes benign peripheral nerve sheath tumors (PNSTs), malignant PNSTs (MPNSTs), hereditary or inflammatory neuropathy, posttraumatic neuroma, intraneural ganglion or other secondary nonneurogenic malignancies such as neurolymphoma and other intraneural epithelial or mesenchymal tumors ( Table 1 ).
| Clinical Features | Imaging Features | |
|---|---|---|
| Neurogenic Lesions | ||
| Benign PNST | ||
| Neurofibroma | Young to middle age Solitary slow-growing lesion Incidental discovery Mild to moderate sensory or motor symptoms and signs Isolated or associated with neurocutaneous syndromes (can occur at early age) | Continuity with nerve (tail sign) and/or multiple fascicles, can be centrally located Target sign Split fat sign Fascicular sign Unencapsulated Homogeneous or targetoid enhancement High ADC (min) on DTI >1.1–1.2 × 10 −3 mm/s 2 Low SUV max (<2–3) on early and delayed F 18 FDG-PET scan |
| Schwannoma | Young to middle age Solitary slow-growing lesion Incidental discovery or painful lesion Mild to moderate sensory or motor symptoms and signs Isolated or associated with neurocutaneous syndromes (can occur at early age) | Continuity with nerve (tail sign) and/or 1 or 2 fascicles, can be eccentrically located Target sign Split fat sign Fascicular sign Encapsulated Cystic changes, hemorrhage, calcification (10%–20%) Homogeneous or targetoid or heterogeneous enhancement High ADC (min) on DTI >1.1–1.2 × 10 −3 mm/s 2 Low SUV max (<2–3) on early and delayed F 18 FDG-PET scan |
| Perineurioma | Young to middle age Isolated Slowly progressive mononeuropathy with few or no sensory symptoms or signs | Continuity with nerve (tail sign) and/or multiple fascicles Most commonly in lower limbs, and in sciatic or femoral distributions Uniform homogeneous fascicular enlargement and hyperintensity (Honeycomb pattern) High ADC (min) on DTI >1.1–1.2 × 10 −3 mm/s 2 Low SUV max (<2–3) on early and delayed F 18 FDG-PET scan |
| Malignant PNST | New-onset sensory and/or motor deficit New or intensified pain Rapid enlargement of a known PNST | Irregular or round shape Usually larger than 5 cm Perilesional edema Intratumoral nodularity, necrosis/cystic change or hemorrhage T1 heterogeneity and lack of targetoid appearance Heterogeneous enhancement Restricted diffusion: low ADC (min) on DTI <1.1 × 10 −3 mm/s 2 Increased SUV max (>3–4) on early and delayed F 18 FDG-PET scan |
| Neurocutaneous syndromes | Younger age of presentation and family history | — |
| NF1 | Cutaneous lesions Skeletal deformities Glioma Lisch nodule First-degree relative | Plexiform neurofibroma or multiple PNSTs (usually NF) Multifocal diffuse nerve thickening(s) and nodularity Malignant PNST |
| NF2 | Vestibular tumors Ependymomas with other schwannomas or multiple meningiomas First-degree relative | Multiple benign PNST (usually SW) Malignant PNST (very rare) |
| Schwannomatosis | No vestibular tumors (rare reports of unilateral vestibular schwannoma) Possible meningioma Multiple PNST | Multiple benign PNST |
| Tumor Mimics | ||
| Neuroma | Prior trauma or surgery | Fusiform shape Spindle (neuroma in continuity) or end-bulb neuroma (complete nerve transection) Heterogeneous T1 and T2 signal alterations with loss of fascicular continuity Lack of significant enhancement Perineural scarring |
| Lipoma | Mononeuropathy with gradual-onset motor and/or sensory symptoms Most commonly in median nerve distribution Macrodactyly and/or association with macrodystrophia lipomatosa | Shape: fusiform or rounded. Diffuse long segment or, less commonly, focal nerve enlargement Fatty and fibrous infiltration with typical MR signal characteristics Spaghetti appearance on long-axis images and coaxial-cable appearance on short-axis images |
| Hereditary neuropathy | Positive family history Hyporeflexia, high arches Foot deformities Two-thirds CMT 1A Two-thirds with positive PMP22 gene | Symmetric and bilateral nerve enlargement Peripheral appendicular and axial pan plexus involvement Most enlargement with CMT 1A Uniform fascicular enlargement Variable to no enhancement Multiple nerve entrapments may be identified, especially with HNPP variant |
| Inflammatory neuropathy: AIDP/CIDP/MMN | AIDP/CIDP: albuminocytologic dissociation. Increased CSF protein Prognosis: good in AIDP, poor in CIDP. In MMN, prognosis is variable | Diffuse (often asymmetrical) > fusiform enlargement of cauda equina, nerve roots/plexus and peripheral nerves MMN common in upper limbs. CIDP common in lower limbs Mild to moderate enhancement. No focal masses as with neurocutaneous syndromes Skeletal survey and cross-sectional examinations may show underlying malignancy, particularly myeloma association with CIDP |
| Infectious neuropathy: leprosy | Endemic area Neuropathic ankles and wrists Neuropathic acro-osteolysis | Ulnar or peroneal involvement Peripheral nerve enlargement with hyperintensity Abscess formation |
| Nonneurogenic Lesions | ||
| Tumor Mimics | ||
| Intraneural ganglion | With internal joint derangement Slow or fluctuating functional motor loss Prognosis: good with surgical resection of the intra-articular feeding nerve | In close proximity to a joint with evidence of internal joint derangement, such as Baker cyst, paralabral cyst, ganglion cyst Elongated shape Cystic intraepineural mass. Mutiloculated Nerve hyperintensity ± peripheral enhancement |
| Amyloidosis | Pertinent long-standing clinical history of chronic kidney disease, myeloma, chronic infection, and so forth Bilateral sensorimotor symptoms. Gradual deterioration Negative birefringent deposits in the epineurium and other layers of the nerve on Congo red staining | Lower extremity more commonly involved, particularly LS plexus and sciatic nerves Diffuse or, less commonly, focal nerve enlargement with nodularity similar to neurocutaneous syndromes, but less pronounced Variable enhancement |
| Tumors | ||
| Lymphoma | Known hematologic malignancy Painful sensorimotor neuropathy | Can present as adjoining adenopathy with compression or infiltration of the nerves or, less commonly, with primary enlargement of the peripheral nerve with multifocal nodularity and areas of T2 hypointensity Intense enhancement on contrast |
| Intraneural epithelial or mesenchymal tumors | Known malignancy in cases of metastasis In primary epithelioid or mesenchymal sarcoma, gradual mononeuropathy symptoms and/or signs | Evidence of direct invasion but local metastasis or sarcoma Evidence of other metastases Heterogeneous fusiform enlargement of the nerve with intraneural primary sarcoma or granular cell tumor Heterogeneous enhancement with tumors. No significant enhancement with lipoma along with its fatty tissue characteristics |
Neurofibromas account for 5% of all benign soft tissue tumors with 95% occurring sporadically. Schwannomas also account for 5% of all benign soft tissue tumors and 95% also occur sporadically. The remainder of neurofibromas and schwannomas are related to genetic neurocutaneous syndromes, including neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and schwannomatosis (SW).
NF1 is one of the most common autosomal dominant neurogenetic syndromes with well-established clinical criteria ( Box 1 ). Benign PNSTs called neurofibromas are the hallmark of NF1. There are 3 main types of neurofibromas: localized, diffuse, and plexiform, and all can be present in patients with NF1. Plexiform neurofibromas are benign but complex tumors that cause diffuse enlargement of deep nerves such as the plexuses or may present as diffuse cutaneous and nerve thickening. These lesions are classic for NF1. They are difficult to manage clinically because they can cause pain, disfigurement, and neurologic dysfunction and can progress to malignant sarcomas.
NF1:
At least 2 of the following:
- •
More than 6 café au lait macules (>1.5 cm in adults and >0.5 cm in prepubertal individuals)
- •
More than 2 neurofibromas of any type or 1 plexiform neurofibroma
- •
Axillary or inguinal freckling
- •
Optic pathway glioma
- •
Two or more Lisch nodules
- •
An osseous lesion specific for NF1, including sphenoid dysplasia or thinning or pseudoarthrosis
- •
A first-degree relative with NF1 by the criteria listed earlier
- •
NF2:
At least 1 of the following scenarios:
- 1.
Bilateral masses of the eighth cranial nerve consistent with vestibular schwannoma
- 2.
First-degree relative, and:
- a.
Unilateral lesion consistent with vestibular schwannoma in someone <30 years old
- a.
Or:
- b.
At least 2 of the following:
- •
Meningioma
- •
Glioma
- •
Schwannoma
- •
Juvenile cortical cataract
- •
- b.
- 3.
Unilateral lesion consistent with vestibular schwannoma in someone <30 years old
And:
- a.
At least 2 of the following:
- •
Meningioma
- •
Glioma
- •
Schwannoma
- •
Juvenile cortical cataract
- •
- a.
- 4.
More than 2 meningiomas and:
- a.
Unilateral lesion consistent with vestibular schwannoma in someone <30 years old
- a.
Or:
- b.
At least 2 of the following:
- •
Meningioma
- •
Glioma
- •
Schwannoma
- •
Juvenile cortical cataract
- •
- b.
- 1.
schwannomatosis:
- 1.
More than 30 years old and all of the following:
- a.
More than 2 schwannomas (1 with pathologic confirmation)
- b.
No vestibular schwannoma by MR imaging with thin cuts through the internal auditory canals
- c.
No NF2 gene mutation
- a.
Or:
- 2.
More than 1 nonvestibular schwannoma (with pathologic confirmation) and a first-degree relative
NF2 is also an autosomal dominant inherited syndrome that predisposes individuals to multiple tumors of the nervous system. Although intracranial schwannomas, such as vestibular schwannomas, are pathognomonic for NF2, patients with NF2 often have peripheral schwannomas. In contrast, patients with schwannomatosis generally have no intracranial lesions but a preponderance of peripheral schwannomas. Familial schwannomatosis is also thought to be caused by autosomal dominant inheritance, although most patients with schwannomatosis have no known familial occurrence. Patients with schwannomatosis develop peripheral schwannomas involving spinal roots, plexuses, and peripheral nerves. The major clinical manifestation of schwannomatosis is pain, often requiring multiple surgical procedures.
Because these syndromes account for a great number of the lesions detected on peripheral nerve imaging and can be difficult to distinguish from either nontumor causes or nonsyndromic causes, this article reviews the typical imaging appearance of PNST and tumorlike conditions with an emphasis on MRN features that distinguish benign tumors from malignant tumors and from conditions that mimic tumors.
Neurogenic Lesions
Benign peripheral nerve tumors
Neurogenic tumors represent 10% to 12% of all benign soft tissue neoplasms. Benign PNSTs often have distinctive features including location in relation to the neurovascular bundle, clinical history, and intrinsic imaging features. The common neurogenic tumors include neurofibroma, schwannoma, and, rarely, perineurioma. Although neurogenic tumors can be diagnosed on MR imaging, it is difficult to differentiate between the schwannomas and neurofibromas. Perineuriomas are less likely to represent a diagnostic dilemma because of their rarer occurrence, location, and more typical clinical and imaging presentation. Jee and colleagues reported that MR imaging findings can aid in differentiating schwannomas and neurofibromas; however, no single imaging finding or combination allows definitive diagnosis, which is not surprising because they share several features and can be difficult to distinguish pathologically.
Neurofibroma
Neurofibromas are benign PNSTs that arise from Schwann cells, but have multiple additional cell types making up the tumor mass, including neuronal axons, fibroblasts, mast cells, macrophages, perineural cells, and extracellular matrix materials such as collagen. These tumors are inseparable from the normal nerve, and therefore complete surgical excision must include the functional nerve. Neurofibromas can occur both in the context of NF1 and sporadically. They also come in several forms including a localized or solitary form, a diffuse form, and plexiform neurofibroma. One of the complexities is that there is not uniform agreement about the definition of neurofibromas across or even within medical subspecialties. However, using the terms most commonly encountered in radiology, the solitary neurofibroma is the most common type encountered clinically overall. These neurofibromas are also termed nodular neurofibromas and are the variety that is most commonly encountered in patients who do not have NF1. These neurofibromas typically present in young to middle-aged adults with a slow-growing mass. They can be incidentally discovered or patients may present with mild to moderate sensorimotor symptoms.
The diffuse, superficial form involves the subcutaneous tissues and is often seen in the region of the head and neck, but can be anywhere in the body ( Fig. 1 ). Plexiform neurofibromas grow along the nerve sheath spreading the axons as the abnormal cells proliferate and increased extracellular matrix is deposited. They may involve multiple fascicles and branches of nerve and can involve nerves in any region of the body. The most common location for plexiform neurofibromas in patients with NF1 is the trunk including the paraspinal region (41%), followed by the neck/upper trunk (24%) and the extremities (17%). Between 15% and 30% of plexiform neurofibromas are isolated to the head and neck region. Diffuse and plexiform neurofibromas are more likely to be found in the setting of NF1.
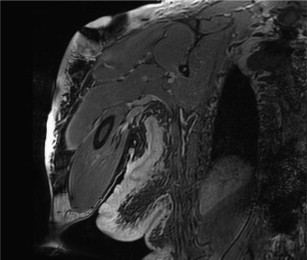
Localized or solitary neurofibromas are often indistinguishable from schwannomas based on imaging features. They are usually well-defined, unencapsulated, fusiform or round soft tissue masses typically less than 5 cm in diameter. The fusiform shape and continuity with the nerve (tail sign) is useful in establishing a neurogenic origin because it reflects the nerve entering and exiting the mass. This morphology is better seen when a large and deep nerve is involved as opposed to small superficial nerve. On T1-weighted (T1W) images, they are usually isointense to muscle. On T2-weighted (T2W) images, they show heterogeneous high signal intensity. The presence of target sign, fascicular sign, split fat sign, and continuity with a peripheral nerve points to a neurogenic origin. These lesions show variable enhancement with gadolinium contrast agent. The target sign refers to high peripheral signal and low to intermediate central signal on fluid-sensitive sequences. The intralesional architecture with more myxoid material peripherally and fibrous tissue centrally accounts for this appearance. Although the target sign was initially thought to be pathognomonic of neurofibroma, it has been observed in both neurofibromas and schwannomas and has even been reported in MPNSTs. The fascicular sign describes multiple ringlike prominent structures within the lesion, possibly reflecting the enlarged fascicular bundles seen histologically. A thin, hypointense capsule might be identified on T2W images, particularly if the tumor is surrounded by fat. This finding is slightly more common in schwannomas than in neurofibromas. On contrast-enhanced images, small nerve sheath tumors often show intense and homogeneous or targetoid enhancement. Large lesions may show predominantly peripheral, central, or heterogeneous nodular enhancement. Specific evaluation for edemalike signal on T2W images, fatty infiltration, and atrophy of innervated muscles is recommended to strengthen the diagnosis of a neurogenic tumor. On advanced diffusion tensor imaging (DTI), these lesions are associated with high apparent diffusion coefficient (ADC) values (>1.1–1.2 × 10 −3 mm/s 2 ).
MR imaging of plexiform neurofibromas shows a tortuous mass of thickened nerve branches that can invade surrounding tissues ( Fig. 2 ). In general, plexiform neurofibromas share the same signal characteristics and enhancement pattern as a solitary lesion. Developing in childhood, plexiform neurofibromas can precede cutaneous neurofibromas. Depending on their location, plexiform neurofibromas can be deep, superficial, or a combination of both. Deep plexiform neurofibromas do not involve the skin or subcutaneous tissues. The superficial plexiform neurofibromas can be asymmetric in distribution, with a diffuse or infiltrative morphology, extend to the skin surface in an arborizing pattern, with smaller fascicles or nodules, and lack a target sign. Superficial neurofibromas can mimic venous malformations on MR imaging, and MR angiography or Doppler ultrasound evaluation may be needed for further differentiation.
Schwannoma
Schwannoma (neurilemoma or neurinoma) is also a tumor of Schwann cells. Schwannomas, unlike neurofibromas, are typically encapsulated and do not have multiple cell types inherent to the tumor. They can be seen at any age but are more commonly diagnosed in patients 20 to 30 years of age. They are usually solitary, slow-growing small lesions (<5 cm) that are incidentally discovered or present with mild to moderate sensorimotor symptoms. They can be found in the cranial, spinal, and sympathetic nerve roots as well as the peripheral nerves of the flexor surfaces of the extremities.
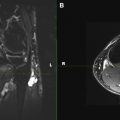
Schwannomas are fusiform masses, eccentric to and separate from the adjacent nerve but encapsulated within the perineurium ( Fig. 3 ). When they arise from cutaneous or other small nerves, the nerve may be obliterated by the mass on MR imaging. They have 2 components: a hypercellular Antoni A region and a loosely organized, hypocellular Antoni B region. Surgical excision of schwannomas, unlike neurofibroma, can spare the affected peripheral nerve. This eccentric and separate relationship of the schwannoma relative to the involved peripheral nerve is not confidently shown on MR imaging to distinguish between neurofibroma and schwannoma. However, dedicated MRN can show the 1 or 2 prominent fascicles individually affected by the tumor, similar to surgical findings. Large tumors may exhibit cystic degeneration, calcification, hemorrhage, and fibrosis, and are described as ancient schwannomas. They only rarely undergo malignant transformation.

Schwannomas share MR imaging features with other neurogenic tumors, especially nodular neurofibromas. A hypointense capsule representing the epineurium is more commonly seen with schwannomas than with neurofibromas. On MR imaging, schwannomas are isointense to skeletal muscle on T1W-images and heterogeneously hyperintense on T2W-images and variable contrast enhancement. Similar to neurofibromas, they can show target sign, fascicular sign, and split fat sign. The target pattern is usually absent in large masses and in tumors with cystic, hemorrhagic, or necrotic degeneration. On advanced DTI, these lesions also show high ADC values or lack of restricted diffusion similar to neurofibromas. Just as neurofibromas can be plexiform, schwannomas can also be plexiform. In general, when evaluating for possible malignant potential in a PNST, heterogeneity in a schwannoma is less concerning than in a neurofibroma, because the latter are generally more homogeneous or simply targetoid.
Perineurioma
Perineurioma are categorized into 2 main types based on location: intraneural or extraneural. Intraneural perineuriomas are benign neoplasms composed of whorls of perineural cells surrounding the peripheral nerves. Because of their histologic origin, they are located in the periphery of the nerves. Cross-sectional histologic examination reveals irregularly enlarged, hypercellular nerve fascicles composed of spindled perineural cells arranged in pseudo-onion bulblike whorls. The mitotic rate is low, accounting for the slowly progressive or static symptoms. The nature of intraneural perineurioma has been debated in the literature, with some investigators speculating a reactive posttraumatic cause. The recent evidence has confirmed its neoplastic nature. These benign tumors are typically seen in teenagers with progressive muscle weakness. Pain and sensory disturbances are less common. As opposed to neurofibroma and schwannoma, which are both associated with good prognosis, perineurioma have poor prognosis with slow functional loss over time.
On MR imaging, the affected peripheral nerve is enlarged, T1 hypointense, T2 hyperintense, with associated avid enhancement on postcontrast imaging ( Fig. 4 ). There is gradual increase of the nerve caliber followed by gradual taper. Fat-saturated postcontrast T1W images are most helpful to show the extent of the lesions. Separating the lesions from adjacent vessels can be difficult and the lesion can be mistakenly characterized as a vascular malformation on imaging. The sciatic nerve or common peroneal nerves are most commonly affected. The fascicular architecture is maintained on axial and longitudinal MRN images. The individual fascicles are uniformly enlarged, resulting in a honeycomb appearance. Although the imaging appearance can mimic other neurogenic benign neoplasms, the clinical history of slowly progressive mononeuropathy, the patient’s young age, and lack of known tumor syndrome makes the diagnosis likely. Because these tumors are static or slowly progressive, they can be followed clinically and with imaging as indicated. Treatment of choice is resection of the lesion with end-to-end nerve grafting. However, few patients do well, despite extensive surgery.
Malignant peripheral nerve tumors
Malignant peripheral nerve tumor (malignant schwannoma, neurofibrosarcoma, malignant neurolemmimas, and neurogenic sarcoma) is a spindle cell sarcoma arising from a peripheral nerve or neurofibroma or showing nerve tissue differentiation. MPNSTs account for 3% to 10% of all soft tissue sarcomas with 15% to 70% occurring in patients with NF1. This sarcoma is a high-grade malignancy with an overall poor prognosis. MPNST more commonly occurs in young persons between 20 and 50 years of age without gender predilection. Patients with NF1 with MPNST present at a younger age and are mostly male. MPNSTs most commonly involve large peripheral nerves including sciatic nerve, brachial plexus, and lumbosacral plexus. Malignant PNST can also be a secondary neoplasm related to previous radiation therapy. Such tumors develop after a long latent period (10–20 years) and account for 11% of malignant PNSTs. Clinical signs and symptoms suggesting an MPNST include new-onset pain, the intensification of existing pain, and the rapid enlargement of a peripheral tumor. Anatomic and metabolic imaging is used in an attempt to distinguish benign neurofibromas in a growth phase from an MPNST.
Using F 18 fluorodeoxyglucose positron emission tomography ( FDG-PET), multiple studies have shown usefulness in differentiation between benign tumours and MPNSTs. In 2008, Ferner and colleagues studied patients with NF1 who had symptomatic neoplasms, using early (60–90 minutes) and delayed (240 minutes) FDG-PET acquisitions, and reported a sensitivity and specificity of 89% and 95% respectively for diagnosing an NF1-associated MPNST. Warbey and colleagues subsequently showed the reproducibility of the early and delayed PET/computed tomography (CT) technique with sensitivity and specificity of 97% and of 87% respectively. Benz and colleagues examined the usefulness of FDG-PET/CT to distinguish histologically proven MPNSTs from benign PNSTs. The difference in standardized uptake values (SUVs) observed in MPNSTs compared with those of schwannomas was not as striking as the difference between SUVs of MPNSTs and neurofibromas, with 3 of 14 schwannomas exhibiting maximal SUV (SUV max ) greater than 5. Such investigations suggest that, although PET is useful for differentiating an MPNST from a neurofibroma, PET is less specific for distinguishing an MPNST from a schwannoma. This limitation is similar to that of anatomic MR imaging, in which imaging features that suggest malignancy, such as large size, necrosis, hemorrhage, and heterogeneity, are similarly observed in schwannomas and MPNSTs.
Using anatomic MR imaging, a few studies have described some distinguishing characteristics for benign and malignant PNSTs. Matsumine and colleagues assessed MR imaging features of neurofibromas and MPNSTs in patients with NF1 and showed that, although irregular tumor shape, indistinct margins, and heterogeneous enhancement were important factors, only intratumoral lobulation and the presence of T1-hyperintense areas were considered diagnostic indicators of malignancy. Four specific MR imaging features were recently reported that can be used to distinguish MPNSTs from neurofibromas with a sensitivity and specificity of 61% and 90% respectively (when 2 or more of the following features were observed: large size, peripheral enhancement pattern, perilesional edema, and intratumoral cystic changes).
Functional and metabolic MR sequences have also been described for the assessment of nerve sheath tumors. Preliminary functional DTI studies in assessing nerve tract integrity in the presence of various peripheral nerve sheath lesions have been performed. Benign PNSTs show near-normal appearance or partial nerve tract disruption in benign tumors, with the exception of degenerated schwannoma and plexiform neurofibroma; however, neurolymphoma, perineurioma, and hereditary neuropathy also showed near-normal appearance of the tracts. Two MPNSTs were also studied using DTI and showed partial and complete tract disruption. Quantitative diffusion-weighted imaging has been reported to be of questionable value for distinguishing benign and malignant soft tissue tumors including PNSTs. In addition, metabolic MR imaging with MR spectroscopy has been studied for the characterization of musculoskeletal lesions for malignancy, and can aid in distinguishing between these two lesions.
Malignant triton tumor (MTT) is a rare subtype of MPNST, histologically defined as a MPNST with additional rhabdomyoblastic differentiation ( Fig. 5 ). The mean age of patients with MTT has been estimated to be 31.7 years. There seems to be an equal sex distribution in those affected, and the tumor coexists with NF1 in 44% to 69% of cases. When associated with NF1, MTT tends to present at a younger age, and in men, than has been observed in sporadic cases. MTT is a highly aggressive tumor with low overall survival rates (estimated 5-year survival rates are 26%), high rates of metastasis (48%), and local recurrence (43%).
Tumor syndromes
The neurofibromatoses are hereditary syndromes caused by genetic mutations in the NF1 , NF2 , and SMARCB1 tumor-suppressor genes, respectively. NF1, NF2, and schwannomatosis increase the likelihood to develop Schwann cell tumors. These related syndromes have overlapping clinical features making clinical and imaging differentiation difficult in some cases; however, there are established diagnostic criteria that assist in reliable clinical differentiation (see Box 1 ).
Despite the benign histology of neurofibromas and schwannomas, they can cause significant morbidity and even mortality. Patients with NF1 have increased mortality caused by MPNSTs, central nervous system gliomas, cardiovascular disease, and organ compression by neurofibromas. Even the benign tumors of NF1 (neurofibromas and optic pathway gliomas), NF2 (schwannoma, ependymoma, and meningioma), and schwannomatosis (schwannoma) increase morbidity because of continuous tumor growth and frequent surgical inaccessibility. Pain is a debilitating consequence of schwannomatosis. Patients suffering from schwannomatosis tend to be younger than those presenting with solitary schwannomas. Therefore, schwannomatosis should be suspected in young individuals presenting with multiple schwannomas but not meeting the criteria for NF2. Increased growth and new pain in a known benign PNST in the context of tumor syndrome should raise suspicion of malignant transformation.
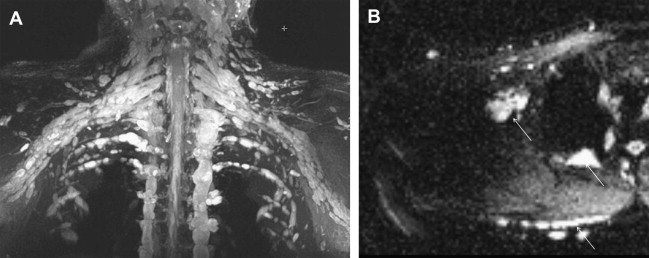
NF1 and NF2 as well as schwannomatosis occur in mosaic forms caused by somatic mutations ( Fig. 6 ). Early somatic mutations cause generalized disease, clinically identical to nonmosaic forms. Later somatic mutations cause localized or segmental. In individuals with mosaic or localized manifestations of NF1, disease manifestations are limited to the affected area. Distribution is usually unilateral but can be bilateral, either in a symmetric or asymmetrical arrangement. Individuals with the mosaic form, even with a generalized phenotype, typically have milder disease and decreased risk for transmission of the gene to their children than individuals with the nonmosaic form, although the risk cannot be accurately quantified.
At present, localized MRN examinations are performed on these patients based on concerning symptoms and physical examination findings. However, these studies inadequately assess whole-body tumor burden and make tracking tumor behavior for tumors that cross multiple anatomic regions more difficult over time. In large centers managing these tumor syndromes, there is a move toward whole-body MR imaging with fluid-sensitive and anatomic imaging. Such a whole-body examination can provide detailed information regarding whole-body tumor burden including the number, distribution, type (localized or plexiform), and size of nerve sheath tumors in a patient. It can also be used in monitoring tumor growth as well as response to treatment.
PNST mimics
Neuroma
Posttraumatic neuroma, also known as neuroma, is related to disorganized proliferation of axonal tissue as a response to injury such as chronic friction or transection. Pain is the most common clinical symptom and can be reproduced with tapping on the lesion (Tinel sign). A soft tissue mass that is firm at a focal pressure site may be apparent. The most common location for traumatic neuroma is the lower extremity after amputation. They can be divided into 2 major categories: neuroma in continuity (NIC) or end-bulb neuroma. NIC is a localized, fusiform enlargement caused by a fibroinflammatory response to chronic friction or irritation. NIC is subdivided into 2 pathologic types: spindle neuromas with intact perineurium or lateral neuromas with partially disrupted perineurium. End-bulb neuromas result from entangled regenerating axons attempting to reestablish continuity after an injury or surgery (amputation). They can develop within 1 to 12 months of the traumatic event and can present as a painful mass.
MR images show fusiform (NIC) or bulbous (end-bulb neuroma) masses in continuity with the injured or transected nerves, respectively. Most traumatic neuromas are of intermediate signal intensity on T1W images and of intermediate to heterogeneously high signal intensity on T2W images with loss of fascicular continuity ( Fig. 7 ). Contrast enhancement is variable. NIC has a similar fusiform morphology to previously described PNST; however, presence of perineural scarring, lack of split fat or target sign, and usually lack of significant enhancement aids in distinguishing the two entities.
Initial nonoperative treatment has been successful in up to 50% of patients. Failure of nonoperative therapy may require surgical resection. Prognosis can be variable depending on the extent of nerve circumference involvement.
LIPOMA/neural fibrolipoma
Lipoma can involve the nerve intraneurally or perineurally. Neural fibrolipoma (lipomatosis or nerve or fibrolipomatous hamartoma) refers to a tumorlike process with intraepineural as well as perineural infiltration of fatty and fibrous tissue. It presents before the age of 30 years, and most commonly affects the median or ulnar nerve in the wrist. The lesions can be associated with macrodactyly in the hand or foot. The slow-growing mass can cause increased pain, paresthesia, and weakness.
MR imaging is pathognomonic in lipoma with simple fatty component in close proximity to the nerve. Neural fibrolipoma shows diffuse enlargement of the affected nerve ( Fig. 8 ). The fatty and fibrous infiltration causes a cablelike appearance of the thickened fascicles ( Fig. 9 ). There is a typical spaghetti appearance on the long-axis images and a coaxial-cable appearance on the axial images. The signal intensity of the tissue surrounding the nerves is variable, related to the relative amounts of fat and fibrous tissue encompassing the lesion.
Hereditary neuropathy
Charcot-Marie-Tooth (CMT) syndrome is a rare hereditary motor and sensory neuropathy that can have a confounding MR appearance imitating tumor syndromes such as neurofibromatosis. A positive family history is present in 80% of patients. The typical clinical presentation includes chronic degeneration of peripheral nerves and nerve roots with predominantly distal muscle atrophy and sensory impairment, hyporeflexia, and foot deformities such as high arch and pes cavus. Two-thirds of these lesions are CMT type 1A (demyelinating type) and two-thirds are positive for PMP22 gene.
Bilateral peripheral nerve and lumbosacral plexus involvement are typically present. Typical MRN findings include fusiform T1 hypointense and T2 hyperintense masslike symmetric enlargement of the peripheral nerves and/or cauda equina ( Fig. 10 ). Type II (axonal variety) shows minimal nerve enlargement but abnormal hyperintensity is also observed. Some of the hereditary neuropathy can show abnormal contrast enhancement. Multiple nerve entrapments can be identified, especially with the type III (hereditary neuropathy with liability to pressure palsies [HNPP]) variant.
Treatment is usually conservative with surgery reserved for orthopedic deformity correction or release of disabling or painful nerve entrapments. Prognosis is poor with slow progression over time.
Inflammatory neuropathy
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a chronic (greater than 8 weeks) analog to Guillain-Barré syndrome. The patients present with muscle weakness, sensory involvement, and areflexia. Lumbar puncture reveals albuminocytologic dissociation with high cerebrospinal fluid protein. Acute inflammatory demyelination polyneuropathy (AIDP) or Guillain-Barré is the acute form with a better long-term prognosis. AIDP is usually diagnosed clinically with history of prior infection, typical physical examination findings, and lumbar puncture. Multifocal mononeuropathy (MMN) is an additional inflammatory condition with a variable prognosis. CIDP is more common in the lower extremity, whereas MMN is more common in the upper extremity.
MRN findings include focal or diffuse, asymmetric fusiform enlargement of cauda equina, nerve roots/plexus, and peripheral nerves ( Fig. 11 ). The affected nerves are T1 isointense, T2 hyperintense with mild to moderate contrast enhancement. Although the diagnosis is typically made on clinical grounds, many patients have atypical presentation requiring imaging and nerve biopsy for confirmation. Skeletal survey and cross-sectional studies can aid in detection of underlying malignancy, particularly myeloma, which can be seen in association with CIDP.
Related posts:
 MR Neurography
MR Neurography
 High-Resolution Magnetic Resonance Neurography in Upper Extremity Neuropathy
Magnetic Resonance Neurography Research
High-Resolution Magnetic Resonance Neurography in Upper Extremity Neuropathy
Magnetic Resonance Neurography Research
 Magnetic Resonance Neurography of the Pelvis a nd Lumbosacral Plexus
Magnetic Resonance Neurography of the Pelvis a nd Lumbosacral Plexus
 High-Resolution Magnetic Resonance Neurography in Upper Extremity Neuropathy
High-Resolution Magnetic Resonance Neurography in Upper Extremity Neuropathy
 Peripheral Nerve Surgery
Peripheral Nerve Surgery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


