Fig. 1
Mechanism of action of Gonadotropin-releasing hormone agonists (GnRH-agonists) on the hypothalamic-pituitary-ovarian axis in the presence of uterine fibroids
Three GnRH agonists are currently approved by the Food and Drug Administration for the treatment of endometriosis in the United States: leuprolide acetate, goserelin acetate, and nafarelin acetate. They differ with respect to dose and mode of administration. Leuprolide comes in a depot form and is administered intramuscularly at a dose of 3.75 mg monthly or 11.25 mg every 3 months. Goserelin can be administered as a monthly 3.6 mg subcutaneous implant or a 3 month 10.8 mg implant. Nafarelin is an intranasal spray, with 200 mcg administered per pump; the daily dose ranges from 400 to 1600 mcg.
Most adverse effects of GnRH-agonist therapy are related to hypoestrogenemia and include vasomotor symptoms, insomnia, and urogenital atrophy (vaginal dryness, urinary urgency and frequency, and painful intercourse). Other common side effects include headache, decreased libido, irregular vaginal bleeding, depression, arthralgia, myalgia, irritability, fatigue, and decreased skin elasticity (Table 1) (Olive 2008).
Table. 1
Adverse effects of gonadotropin-releasing hormone agonist therapy
Common (>60 % of patients) |
Hot flashes |
Less common (20–60 % of patients) |
Headache, insomnia, memory disorder, and significant temporary bone mineral density loss (if used 6 months or less) |
Infrequent (2–19 % of patients) |
Significant and persistent bone mineral density loss, anxiety, dizziness, asthenia, depression, vaginal dryness, dyspareunia, weight change, arthralgias, myalgias, alopecia, peripheral edema, breast tenderness, irritability and fatigue, decreased skin elasticity, decreased libido, nausea, altered bowel function, and irregular vaginal bleeding |
Rare (<2 % of patients) |
Vaginal hemorrhage, allergic reactions |
The most worrisome effect of the drug is loss of bone mineral density, which occurs at a rate of roughly 6 % annually (Hornstein et al. 1998). This side effect is largely responsible for the hesitancy to use GnRH agonist for extended periods of time. Another concerning side effect is memory impairment, which effects up to 44 % of those treated. Fortunately, this adverse phenomenon is completely reversible with discontinuation of the drug (Newton et al. 1996; Sherwin and Tulandi 1996).
An adverse effect unique to the use of these drugs for the treatment of fibroids is significant vaginal hemorrhage roughly 5–10 weeks after initiation of medication. This event is due to degeneration and necrosis of submucous myomas, and occurs is approximately 2 % of treated women (Friedman 1989).
The long-term side effects of GnRH agonists can be minimized or eliminated by adding back a small amount of sex steroids, similar to the replacement hormone dosages for menopausal women. A number of different regimens have been utilized, including estrogen/progestin, progestin alone (medroxyprogesterone or norethindrone), and androgen (tibolone). All reduce vasomotor symptoms, but progestins derived from progesterone (such as medroxyprogesterone) tend to preserve bone less well than norethindrone, whose breakdown products include ethinyl estradiol (Chwalisz et al. 2012). Such therapy can begin with the initiation of the medical therapy, or can be delayed for a period of time to allow for unopposed GnRH action.
GnRH agonists are most commonly used as a preparatory treatment for surgery. When treated for at least 3 months, uterine size is reduced to 30–65 % (Friedman et al. 1988; Carr et al. 1993; Minaguchi et al. 2000). Symptomatic relief is often achieved during this time, as bleeding is generally reduced and pressure effects relieved (Benagiano et al. 1996). When patients are anemic from excessive uterine bleeding, the use of GnRH agonist preoperatively has been a strategy used to allow increase in hemoglobin levels. Furthermore, this approach often decreases blood loss at surgery. However, one caveat often mentioned by surgeons (but heretofore undocumented in the literature) is that pretreatment softens the myomas, making complete removal more difficult.
For the hysteroscopic removal of submucous myomas, GnRH pretreatment has several particular advantages. First, when removing fibroids by pieces (as with a resectoscope) rather than as a single, intact entity, a reduction in volume becomes much more meaningful. Second, endometrial atrophy will improve visualization. Finally, decreased vascularity should minimize blood loss and fluid intravasation.
It must be kept in mind that the effect of GnRH agonist reduces leiomyoma cell size and is not cytotoxic. With cessation of the drug there is a return in fibroid size within 3–4 months. This would imply that its use as a stand-alone therapy would be extremely limited. One such situation would be the use of GnRH agonist to treat women close to menopause in an attempt to avoid surgical intervention. However, intermittent, long-term use in women with fibroids may be feasible; in a study of women treated with the medication for abnormal uterine bleeding attributable to fibroids, there was symptomatic improvement in half for up to 6 months following discontinuation (Scialli and Levi 2000). Further investigation is clearly needed to evaluate this interesting concept.
4.1.2 GnRH Antagonists
GnRH antagonists are analogs of the GnRH molecule that act by directly competing for and occupying pituitary GnRH receptors. This blocks access of the GnRH molecule to these receptors, resulting in immediate pituitary suppression of gonadotropin secretion. This avoidance of the initial gonadotropin flare seen with GnRH agonists allows the antagonist to cause a clinical effect much more quickly, generally within 2 weeks. It is also rapidly reversible with discontinuation of the drug.
Similar amounts of shrinkage of fibroids are seen with antagonists as with agonists (Felberbaum et al. 1998, 2001; Gonzalez-Barcena et al. 1997; Flierman et al. 2005). Currently, long-acting antagonists are not available in the US, but should they become available this drug class will likely supplant GnRH agonists as a preoperative therapy, due to the more rapid onset. Exactly this pattern is seen in countries where GnRH antagonists are available and have begun to cause a decline in usage of GnRH agonists.
5 Selective Estrogen Receptor Modulators
Selective estrogen receptor modulators (SERMs) are nonsteroidal estrogen receptor ligands that act as estrogen agonists in some tissues and as receptor antagonists to block estrogen action in others. Different SERMs behave differently in each tissue in which they act; the specific action is determined by their structure, the estrogen receptor to which they bind, and other interacting molecules in the tissue. As an example, tamoxifen and raloxifene, two well-known SERMs, both have estrogen antagonist effects upon mammary tissue. However, raloxifene is also an estrogen antagonist in endometrium, whereas tamoxifen is an agonist in uterine tissue.
The ability to block estrogen action means that this class of drugs has a potentially therapeutic effect in the treatment of fibroids. However, it is critical to find the SERM with the correct combination of actions in other locales. Tamoxifen has not been used in the treatment of myomas because of its tendency to produce endometrial hyperplasia (Neven et al. 1989). In addition, case reports suggest that tamoxifen may be agonistic in myometium and fibroids as well. Conversely, raloxifene’s agonist/antagonist profile seems much better suited to this purpose.
As should be expected, the side effect profile of a SERM is primarily dependent upon its actions in various tissues. For raloxifene, the principal side effect is vasomotor symptomatology, as the antagonistic effect predominates in the central nervous system. The most critical side effect, however, is the thrombogenic nature of this medication, an estrogen-like activity in the liver that raises the risk of a thromboembolic event threefold (Ettinger et al. 1999). For this reason, leg pain and respiratory complaints must be taken seriously in women treated with this drug, and it is likely contraindicated in women with known thrombophilias.
Initial studies with raloxifene demonstrated that 60 mg daily was sufficient to significantly decrease the size of the tumors for up to a year in postmenopausal women (Palomba et al. 2001). However, in premenopausal women, this dose was ineffective; doses as high as 180 mg daily were required to even see a marginal response in some women (Palomba et al. 2002a). It appears that raloxifene is capable of blocking the effect of estrogen on fibroids when levels are relatively low, but when premenopausal levels are present the blockade is ineffective.
The conflicting data from pre-and postmenopausal women led to the notion that raloxifene might be efficacious if premenopausal women could be “converted” via a medical menopause. Palomba and colleagues combined raloxifene with GnRH agonist treatment in an attempt to do just that (Palomba et al. 2002b). A randomized trial comparing GnRH agonist plus raloxifene 60 mg daily versus GnRH agonist plus placebo demonstrated that after 6 months substantially more fibroid volume reduction was seen with combination therapy than the control arm (Fig. 2). The size reduction remained stable for an additional 12 months of treatment, and myoma symptoms also remained improved (Palomba et al. 2004). The primary side effect, as expected, was hot flashes, but endometrium remained atrophic and there was no significant decrease in bone mineral density. Thus, the combination therapy proved superior in fibroid treatment to either drug alone, and also ameliorated a major adverse effect of GnRH agonists by protecting bone density. This regimen may prove to replace solo GnRH agonist therapy when medical therapy is desirable.
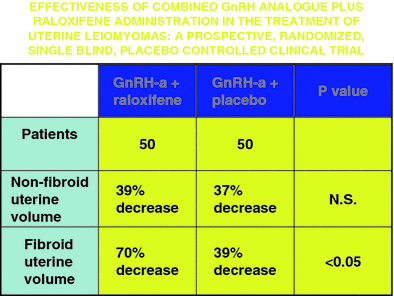
Fig. 2
The effect of GnRH-agonist plus raloxifene versus GnRH-agonist alone on uterine volume and leiomyoma volume. Data from Palomba et al. 2002b
6 Aromatase Inhibitors
Aromatase inhibitors are molecules that directly inhibit estrogen synthesis by either blocking or inactivating aromatase, the enzyme responsible for synthesizing estrogens from androgen precursors. While estrogen is principally produced in the ovary premenopausally, the aromatase enzyme can be found elsewhere, such as in adipose tissue and endometriosis. Aromatase is also produced in many fibroids, which may explain why some tumors do not regress in hypoestrogenic states (Ishikawa et al. 2009).
Given the above facts, it would be anticipated that aromatase inhibitors would be of value in pre- and postmenopausal women. Side effects in such patients would be those expected in a hypoestrogenic state: Hot flashes, vaginal dryness, and bone loss. However, the utility in women with functioning ovaries might be expected to be poor, in that a hypoestrogenic state would likely stimulate the hypothalamic-pituitary-ovarian axis, with greater levels of FSH production, and resultant follicular development; such developing follicles would be unable to ovulate due to an absent LH surge (as estrogen, the surge trigger, is low) and ovarian cysts would result.
Data are limited for the use of these agents in the treatment of fibroids. Most publications are case reports or small uncontrolled series. However, there is currently one randomized trial comparing the aromatase inhibitor letrozole 2.5 mg/day to GnRH agonist in the treatment of uterine fibroids (Parsanezhad et al. 2010). In this study, premenopausal women with a single fibroid >5 cm in diameter were treated for 12 weeks. The patients treated with letrozole had 45 % reduction of volume of the fibroid, a result that did not differ from the GnRH agonist group. However, the patients treated with letrozole had a more rapid onset of size reduction, saw no changes in steroid hormone or gonadotropin levels, and also had no development of ovarian cysts. The long-term or preoperative use of their medication may well prove superior to the existing medications.
7 Progesterone Receptor Modulators
As progesterone has been shown to be critical to the growth of fibroids, a logical pharmaceutical approach to the treatment would be better to inhibit the action of progesterone on these tumors. Progesterone receptor modulators are a class of compounds that interfere with the progesterone–progesterone receptor interaction. Two types of such inhibitors have been developed: (1) the pure progesterone receptor antagonist, and (2) the selective progesterone receptor modulator, which has either agonist or antagonist activity depending upon the site of action.
7.1 Progesterone Antagonists
Mifepristone is a progesterone receptor antagonist with little or no agonist activity. It binds to the progesterone receptor with high affinity, and also binds to the androgen and glucocorticoid receptors. The drug has been shown to reduce the progesterone receptor number in fibroids (Bouchard et al. 2011).
Side effects of this medication include vasomotor symptoms, nausea, and fatigue, but all are relatively infrequent. High doses can produce anti-glucocorticoid effects, and thus have been avoided in recent trials. The most concerning adverse effect is the effect of the drug on the endometrium. The inhibition of progesterone action on the endometrium creates a milleu of pure estrogen stimulation, resulting in histologic changes resembling hyperplasia. This was noted in 28 % in one of the original studies (Steinauer et al. 2004), although re-evaluation of the biopsies later reduced this rate to 14 % (Eisenger et al. 2003). Nevertheless, this finding raises concern for the value of progesterone antagonists in the long-term treatment of uterine fibroids.
Mifepristone has been shown to reduce fibroid volume consistently, by around 50 % in most studies with a dosage range of 5–50 mg/day (Steinauer et al. 2004). The studies also report a high rate of symptom relief (up to 75 %) and a 91 % rate of amenorrhea. Recently, a randomized trial comparing 5 and 10 mg daily doses for 6 months again demonstrated significant volume reduction, but also found that 1-year posttreatment in the majority of women remained asymptomatic (Esteve et al. 2012). Also interesting is an open-label study of the utility of 2.5 mg mifepristone daily. While the uterine volume was reduced only 11 % over the 6-month trial, the improvement in quality of life was similar to that seen with higher doses (Eisinger et al. 2009). This raises the possibility that effective medical treatment may not hinge upon reducing fibroid size in many women.
Ulipristal is a pure progesterone antagonist with no agonist activity. It binds progesterone receptors, but not estrogen receptors, and has less antiglucocorticoid activity than mifepristone (Hild et al. 2000; Attardi et al. 2000, 2002). The drug has been shown to reduce progesterone receptors, and downregulate growth factors and their receptors in cultured leiomyoma cells (Wang et al. 2006; Xu et al. 2006).
The side effect profile of ulipristal shows it to be extremely well tolerated. Estrogen levels are maintained in the mid-follicular range, and thus vasomotor symptoms are infrequent and less profound. There is no evidence of effect upon bone mineral density, and anti-glucocorticoid effects seem to be rare. The effect on the endometrium, a major concern for these medications, has been thoroughly investigated. Over 60 % of women treated with ulipristal show changes typical of this class of drugs after 3 months: Glandular dilatation and dyssynchronous glands and stroma. However, these changes resolved after a 6-month drug-free period (Donnez et al. 2012a). This leads to the intriguing possibility that intermittent therapy might eliminate the concerns of endometrial hyperplasia with this medication.
A number of small studies had previously suggested the value of ulipristal in the treatment of myomas, but two recent randomized clinical trials highlighted the value of this drug. In one, a randomized trial compared ulipristal 5 and 10 mg daily to placebo preoperatively in women with anemia (Donnez et al. 2012b). Bleeding was controlled in over 90 % of those treated with active medication, and there was a significant (but small) decrease in fibroid volume. A second trial compared ulipristal to a GnRH agonist. Compared to the GnRH agonist, ulipristal-induced amenorrhea more rapidly and produced a greater increase in hemoglobin levels in treated patients. While GnRH agonist produced a greater size reduction in the fibroids, the effect of ulipristal appeared to be more sustained. This had been suggested in prior studies (Eisenger et al. 2005).
Thus, ulipristal appears to be a drug worthy of consideration in the preoperative treatment of uterine fibroids; furthermore, its value as a long-term medical treatment via intermittent use is conceivable, and is in need of investigation.
7.2 Selective Progesterone Receptor Modulators
As stated earlier, some progesterone receptor antagonists have agonistic properties in some tissues, organs, or hormonal milleus. These medications are termed selective progesterone receptor modulators, or SPRMs. The most intensely investigated of this class is asoprisnil. This medication exhibits a mixture of agonist and antagonist actions, with a high degree of uterine selectivity. It induces amenorrhea and suppresses endometrial growth (Chwalisz et al. 2006). In addition, it has no antigluticorticoid effects. In culture, asoprisnil produces an anti-proliferative effect upon cultured leiomyoma cells (Chen et al. 2006).
Phase 1 trials with asoprisnil demonstrated reversible suppression of menstruation. Phase 2 trials showed, via multicenter randomized trials, that the drug reduced fibroid size by 36 % and menorrhagia in a dose-dependent manner, with an amenorrhea rate of up to 83 % with a dose of 25 mg daily (Chwalisz et al. 2007). The drug has also been shown to reduce uterine artery blood flow (Wilkens et al. 2008).
Asoprisnil appeared to have relatively few and mild side effects in the above-mentioned trials; however, endometrial histologic changes were seen typical of progesterone receptor antagonists. As a result, phase 3 trials of the drug were abandoned despite the encouraging results.
8 Other Available Therapies
8.1 Danazol
Danazol is an isoxazole derivative of 17-alpha-ethinyltestosterone, and functions in a number of different ways that may inhibit the growth and maintenance of uterine fibroids. The drug alters GnRH pulsatility, resulting in ovulation dysfunction (Panidis et al. 1994), and also creates hypoestrogenemia by inhibiting multiple enzymes in the steroidogenic pathway, including aromatase (Barbieri and Ryan 1981; Ishihara et al. 2003) (Fig. 3). Furthermore, by binding to SHBG, testosterone is displaced and the amount of free testosterone rises. This combination of anti-estrogenic, anti-progestogenic, and androgenic effects seems to be beneficial in reducing fibroid size. Additionally, danazol appears to increase the uterine artery impedance to blood flow.
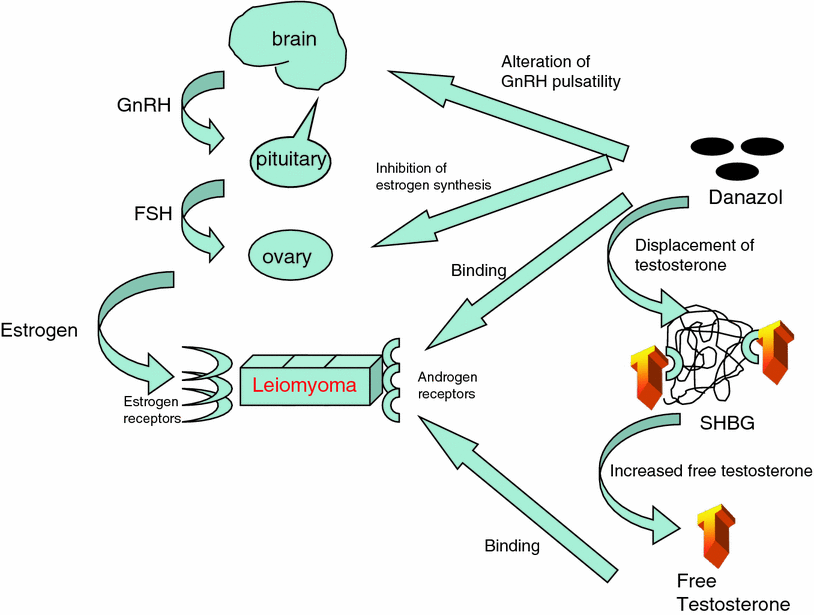
Fig. 3
Mechanism of action of danazol upon uterine fibroids
Clinical studies suggest that the medication is efficacious while being administered, and perhaps for a period of time thereafter. Danazol 400 mg/day produced a 24 % decrease in uterine volume by 4 months; the size was still significantly reduced 6 months after discontinuation of treatment. Symptom reduction was noted in all women treated (De Leo et al. 1999). A lower, more tolerable dose of the androgen has also been tried; women were treated with 100 mg daily for 6 months resulting in a mean myoma volume decrease of 38 % (La Marca et al. 2003).
Unfortunately, danazol has multiple androgenic side effects such as weight gain, acne, inappropriate body hair growth, oily skin, and decreased breast size. These and other less frequent symptoms may be reduced or eliminated, however, with lower doses of the drug, and trials of 50–100 mg daily for extended lengths of time are currently being planned.
8.2 Cabergoline
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


