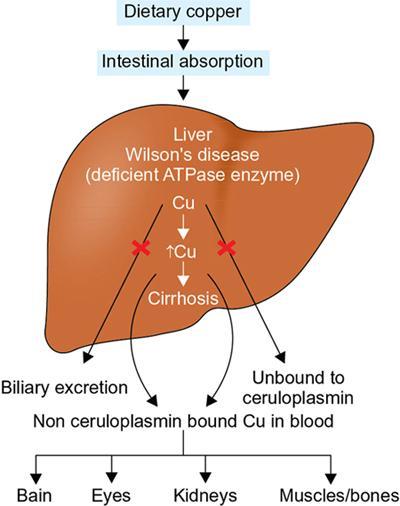
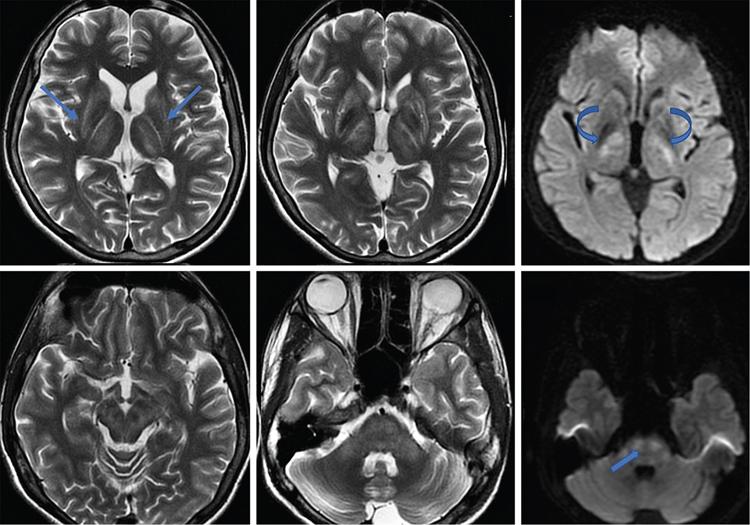
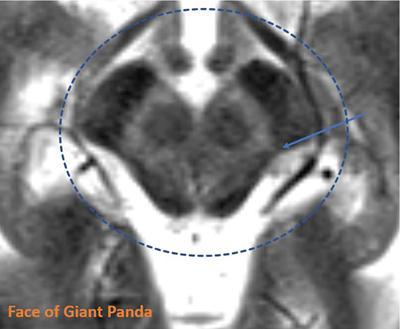
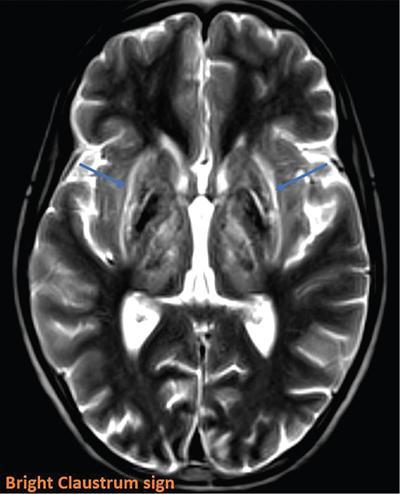
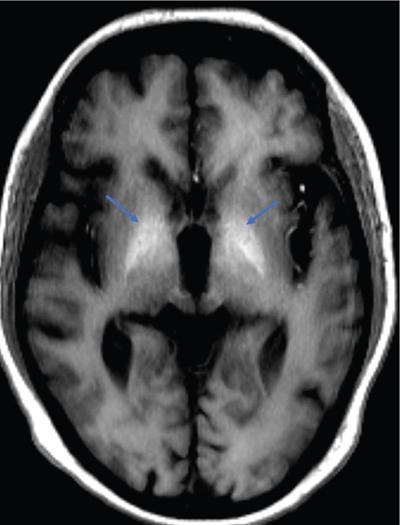
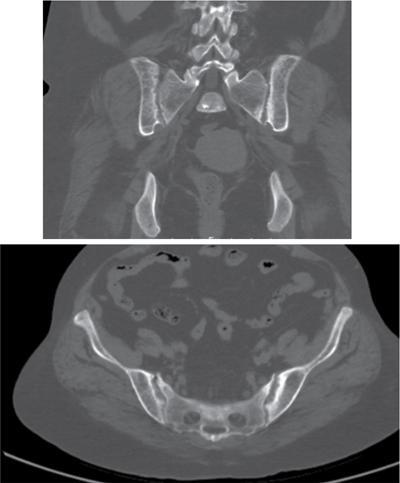
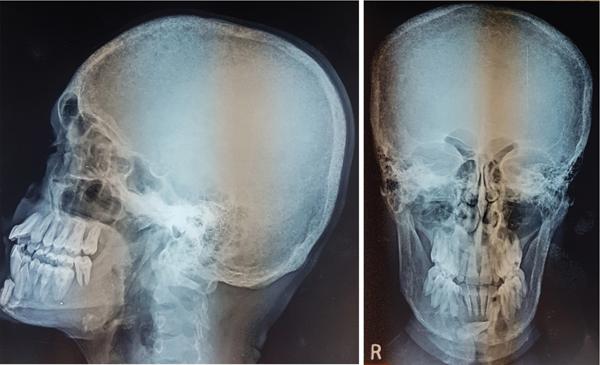
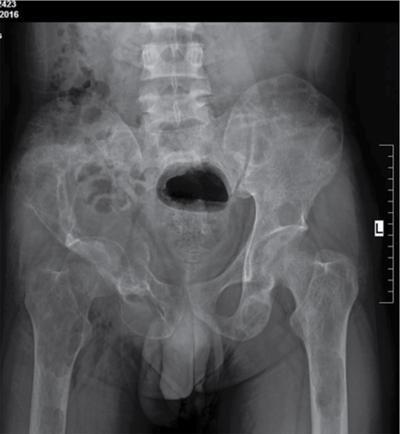
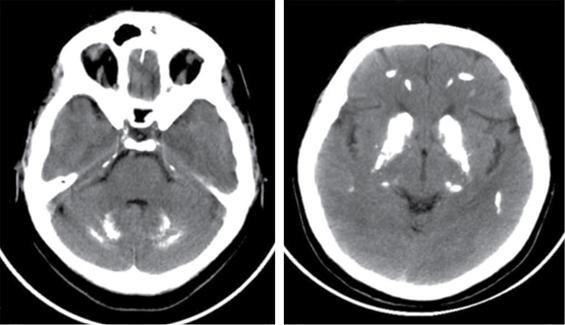
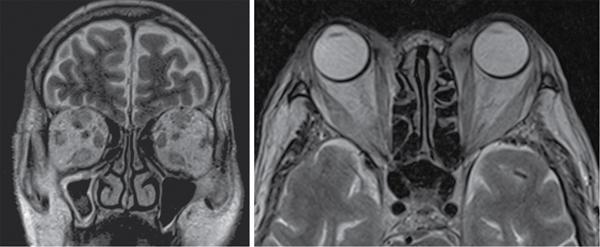
Harsha Chadaga Metabolic bone diseases are a group of diseases that result in abnormalities of (a) bone mass, (b) structure mineral homeostasis (c) bone turnover or (d) growth. Endocrine disorders are a result of malfunction of endocrine systems which could be the gland, hormones, receptors or organs impacted by the hormones. These disorders can cause wide ranging effects on the body. These disorders usually arise from effects of decreased or increased secretions of the hormones. A metabolic disorder occurs when an improper level of a hormone alters the body metabolism and impacts its function. Wilson disease is a disorder of copper metabolism which most often manifests in children and adolescents due to copper accumulation in the liver and brain resulting in hepatic dysfunction and neuropsychiatric symptom. Osteomalacia is the result of impaired mineralization of newly formed osteoid, which leads to characteristic Looser zones and bone softening. Findings of hyperparathyroidism are the result of bone resorption, most often manifesting as subperiosteal resorption in the hand followed by other parts of skeletal system. Renal osteodystrophy is a constellation of skeletal changes seen in patients with chronic renal failure and associated with changes of secondary hyperparathyroidism and can also demonstrate features of ‘rugger jersey spine’, osteomalacia and osteosclerosis. Hypoparathyroidism is most frequently due to iatrogenic injury, and on radiographs hypoparathyroidism reflects an overall increase in bone mass. Thyroid hormone is involved in regulating endochondral bone formation, and congenital hypothyroidism, if untreated, can result in delayed bone age and absent or fragmented distal femoral and proximal tibial epiphyses. Thyroid acropachy manifests as soft tissue proliferation and is most often observed in the hands and feet. Hemochromatosis refers to the accumulation of iron in the different tissues of body secondary to excess iron in the body due to multiple causes affecting the liver, spleen and skeletal system. The findings of acromegaly are due to excess growth hormone secretion and therefore result in proliferation of the bones and soft tissues. WILSON’S DISEASE Sriram Patwari Wilson’s disease (WD), also known as hepatolenticular degeneration, is an uncommon autosomal recessive disorder of the copper metabolism resulting from mutation of ATP7B gene. The disease was first identified by Samuel Alexander Kinnier Wilson, a British neurologist in 1912. WD is distributed throughout the world. The reported prevalence of WD varies between 12 and 29 per 100,000 in Europeans, and prevalence of 33 and 68 per 100,000 is reported in Asian countries other than India. There are no major community-based epidemiological studies of WD in India. The ATP7B gene is located on chromosome 13q14-21 and encodes an enzyme P-type ATPase essential for synthesis of ceruloplasmin and biliary excretion of excess copper. As a result of mutation on the ATP7B gene, there is progressive accumulation of the copper in the liver tissue, resulting in cirrhosis. The liver also releases the excess copper into the bloodstream that is unbound to ceruloplasmin, which precipitates in the brain (lentiform nucleus), eyes and kidneys (Fig. 2.1.1.1). WD most often manifests in children and adolescents with no specific genetic predilection. Since the most common sites of copper accumulation is the liver and brain, WD symptomatically presents with hepatic dysfunction and neuropsychiatric symptom. The CNS manifestations of WD is due to copper deposition in basal ganglia (especially putamen), midbrain, pons and thalamus. CT: The role of CT is limited with widespread availability of MRI. There can be symmetrical hypodensities in bilateral basal ganglia and thalamus. In patients with long-standing WD, there can diffuse cerebral and cerebellar atrophy. Imaging findings include steatosis, acute hepatitis, cirrhosis and portal hypertension. In long-standing cases due to excessive copper deposition, T2 hypointense and T1 hyperintense nodules can be seen on MRI. Imaging findings include early osteoarthritis, chondrocalcinosis, diffuse osteopenia and secondary osteomalacia due to renal dysfunction. The important biochemical investigations in patients with WD include estimation of serum copper (reduced), serum ceruloplasmin (reduced) and 24-h urine copper (increased). The liver biopsy can be done to estimate the degree of liver parenchymal changes and quantification of copper accumulation. Genetic analysis is often performed in the mutation in ATP7b gene. HYPERPARATHYROIDISM AND HYPOPARATHYROIDISM Harsha C Chadaga Hyperparathyroidism refers to increased production and secretion of parathyroid hormone from the parathyroid gland, of which there are many causes. This hormone causes increased osteoclastic activity causing widespread skeletal changes which is specific enough for radiologic diagnosis. In primary hyperparathyroidism, elevated parathormone stimulates osteoclastic resorption, liberating calcium and phosphorus into the bloodstream. Phosphorus is readily excreted and owing to the constant calcium-phosphorus product, calcium is retained, disturbing homeostasis. This results in hypercalcaemia and hypophosphatemia. In secondary hyperparathyroidism, abnormal renal vitamin D formation and calcium loss creates hypocalcaemia and increases the release of parathormone and bone resorption. The histopathologic alterations consist of osteoclastic and osteocytic resorption, with fibrous tissue replacement. The bone is decreased in density and exhibits defective lamellar structure, producing a soft, fragile bone. The pathological hallmark is subperiosteal bone resorption of the outer cortex at the insertional points of ligaments and tendons. There are three basic forms of hyperparathyroidism: primary, secondary, and tertiary. Some of the syndromic associations include multiple endocrine neoplasia type I, type IIa, familial hypocalciuric hypercalcaemia and few others. Hyperparathyroidism typically present in women 30–50 years of age with weakness, lethargy, polydipsia and polyuria. Renal calculus may be the presentation for examination sometimes. Diffuse bone pains, dementia, depression, constipation and pancreatitis are other clinical features which could be the first presentation. The radiological features can be discussed as Bone softening: Bone softening results in basilar impression of skull, biconcave vertebral deformities with kyphoscoliosis and bowing of long bones. Bone softening also results in slipped capital femoral epiphysis. Brown tumours: Also known as osteoclastoma, it appear as focal osteolytic areas with bone swelling. These are result of osteoclastic activity stimulated by parathormone. In the era previous to dialysis, brown tumours were more common in primary form, but now also seen in secondary. Brown tumours are typically seen in ribs, pelvic bones, metaphysis of long bones and mandible (Fig. 2.1.2.5). These are often expansile solitary lesions located eccentric or cortical with no adjacent reactive bone formation. Osteosclerosis: More frequent in secondary hyperparathyroidism. Role of calcitonin has been implicated in osteosclerosis. This is frequently seen involving axial skeleton, pelvis and clavicle. In the spine typical “rugger jersey spine” appearance is noted secondary to osteosclerosis. Soft tissue calcification: More frequently seen in secondary hyperparathyroidism. Typically seen around hip, knee, shoulder and wrist. Calcification of cartilage in menisci is seen and known as chondrocalcinosis (15%–18%). Calcification is also seen in bilateral basal ganglia, dentate nuclei and subcortical white matter. Manifestations of hypercalcemia will appear as nephrocalcinosis and renal calculi. This is seen up to 75% of hyperparathyroidism patients. Erosive arthropathy: It simulates rheumatoid arthritis with joint space preservation. Periosteal new bone formation linear new bone formation paralleling cortical surface. This is typically seen along ileopectineal line, humerus, femur, tibia and radius. Primary HyperParathyroidism Secondary HyperParathyroidism +++ more common, + less common Although US examination, CT of the neck, and magnetic resonance imaging are useful in the evaluation of primary hyperparathyroidism, 99mTc-sestamibi parathyroid scintigraphy is now the best preoperative localizing modality for the detection of parathyroid adenomas. As parathyroid scintigraphy can be sometimes limited by the coexistence of thyroid nodules or other metabolically active tissues, it is often correlated with CT results to yield functional and anatomic localization. Differential Diagnosis: Differential for hypercalcaemia includes metastasis (second most common cause) that will have low or suppressed parathyroid hormone levels and benign familial hypocalciuric hypercalcaemia in which calcium creatinine clearance ratio is < 0.01. Treatment: Most patients with primary hyperparathyroidism are asymptomatic, treatment is controversial. Some clinicians believe that surgical management is always appropriate given the unpredictable long-term complications including recurrent urolithiasis and osteoporosis. NIH consensus statement in 1990 concluded that surgical intervention is the acceptable treatment for primary hyperparathyroidism, surveillance is justified in patients whose calcium levels are only mildly elevated and whose renal and bone status is close to normal. It occurs due to reduced secretion of parathyroid hormone from the parathyroid glands. Parathyroid hormone is essential in calcium metabolism and helps maintain serum calcium milieu.1 Absent or reduced parathyroid hormone results in failure to mobilize calcium from bone, reabsorb calcium from the distal nephron or activate renal 1 alpha hydroxylase activity. Causes of hypoparathyroidism can be congenital or acquired. Some of the common congenital causes are DiGeorge syndrome, type I autoimmune polyglandular syndrome, X-linked or autosomally inherited hypoparathyroidism and PTH gene mutations. In acquired causes there is damage to the para thyroid glands resulting in secondary hypoparathyroidism. Post-surgical hypoparathyroidism is most common acquired cause, usually following thyroid or other head and neck surgeries. Other acquired causes could be due to hemochromatosis, thalassemia major, Wilson disease or metastases (rarely). Most often imaging features come into light during radiographs done for some other purpose. Radiography is the most commonly used modality to evaluate the skeletal and soft tissue changes of hypoparathyroidism. Osteosclerosis either localized or diffuse is most common skeletal finding seen in hypoparathyroidism. Osteosclerosis mainly involves and manifests as thickening of the facial bones and cranial vault with a widened diploe. Other findings are sutural diastasis, which is due to increased intracranial pressure. There is calcification of ligaments mainly anterior longitudinal ligament and posterior paraspinal ligaments in the spine and muscle insertions (enthesopathy). Hypoparathyroidism also results in hypoplastic dentition, delayed or failed eruption of the teeth, and a thickened lamina dura. Subcutaneous calcifications, mainly seen around shoulders and hips, can be seen as well. There are band-like radiodensities in the metaphysis of long bones, and sclerosis of the iliac crests and the margins of the vertebral bodies. Premature closure of the physis is also radiographic finding usually in the developing skeleton. Intracranial calcifications mainly in basal ganglia, falx, and rarely the cerebellum and choroids plexus are seen and occurs due to the metabolic impairment. These are usually seen on CT of brain and less likely in skull radiographs (Fig. 2.1.2.6). Ultrasound helps in identifying renal findings seen in hypoparathyroidism. On ultrasound there is nephrocalcinosis and is usually seen in the patients receiving treatment due to the resultant calciuria. Role of MRI: MRI is not a primary modality used to diagnose hypoparathyroidism. Findings of calcification of ligaments, osteosclerosis, intra-cranial and soft tissue calcifications are also seen on MRI performed for some other purpose and appear as hypointense signal changes in the involved tissues. PSEUDOHYPOPARATHYROIDISM Praveen Wali It encompasses the disorders secondary to end-organ resistance to PTH, mainly kidneys and skeleton. In kidneys, the resistance is seen at the receptor levels in renal tubules and in skeletal system in the osteocytes and osteoclasts. Renal resistance to PTH can be diagnosed by lab investigations by assessing the levels of nephrogenous cyclic AMP and urinary phosphorous levels which fail to increase after parenteral administration of PTH extract. In skeletal system, PTH regulates calcium homeostasis. With end-organ resistance, this homeostasis is not achieved and results in hypocalcaemia. This cannot be corrected by parenteral injection of PTH extract due to end-organ resistance. Normally, PTH stimulates osteoclasts resulting in bone remodelling in normal skeleton. So the manifestations depend on whether the end-organ resistance is at the level osteoclasts or osteoblasts. If osteoclasts are resistant, then the histologic and radiologic features of osteitis fibrosa will not be observed. If the osteoclasts respond to PTH, features of increased bone remodelling with osteitis fibrosa are seen. Based on variability in end-organ resistance, there are several clinical variants of pseudohypoparathyroidism. Subtypes are based on biochemical results and have no specific imaging findings. Pseudopseudohypoparathyroidism (PPHP) is an inherited disorder and is biochemically normal but phenotypically similar to pseudohypoparathyroidism type 1a, called AHO phenotype. These patients have similar clinical and radiological features as pseudohypoparathyroidism but without alterations in parathyroid hormone levels and calcium metabolism. There is often a family history of pseudohypoparathyroidism. PHP and PPHP, although rare we must be aware of imaging features and must be suspected in cases with any of the above findings. Usually, majority of the findings can be imaged by radiography, whilst the CNS manifestations might need cross-sectional imaging. Imaging features in isolation are not specific and must be evaluated in entirety and along with laboratory findings. HYPERTHYROIDISM AND HYPOTHYROIDISM Harsha C Chadaga Hyperthyroidism refers to increased production and secretion of thyroid hormone from the thyroid gland. Hyperthyroidism is not synonymous with thyrotoxicosis, which refers to a clinical syndrome of excess thyroid hormone. Graves’ disease (also known as Basedow disease) is an autoimmune thyroid disease and is the most common cause of thyrotoxicosis (upto 85%). Pathophysiology: Numerous causes of hyperthyroidism are present which include Clinical presentation: Hyperthyroidism brings a constellation of symptoms which include appetite change, insomnia, fatigue, menstrual issues, hand tremors, heart beat irregularities, bowel disturbances, weight fluctuations and many more. The combination of exophthalmos, palpitations and goiter is called the Merseburger (or Merseburg) triad. The radiological features can be discussed as follows: Skeletal: Which includes diffuse osteoporosis and thyroid acropachy. Thyroid acropachy is seen in 1% of Graves’ disease. Radiographic findings of thyroid acropachy are best seen in the hands and feet, in which clubbing, periostitis and soft-tissue swelling is noted. The periostitis is greatest along the radial margins of the metacarpal, metatarsal and middle and proximal phalangeal diaphysis. Soft-tissue swelling may also be seen in the pretibial region. CNS: Manifests as encephalopathy associated with autoimmune thyroid disease (EAATD). MRI studies are frequently normal (around 60%). When abnormalities are present they are non-specific, such T2/FLAIR subcortical white matter high signal intensities and post-contrast meningeal enhancement can be seen. Most patients respond to gluco-corticoid therapy. Thyroid-associated orbitopathy: It is the most common cause of proptosis in adults. It is characterized by bilateral symmetrical enlargement of extraocular muscle belly giving a “coke-bottle” sign with relative sparing of tendinous insertion at globe. Involvement of the extraocular muscles in decreasing order of frequency – levator palpebrae superioris, inferior rectus, medial rectus, superior rectus, lateral rectus and oblique (Fig. 2.1.4.1).
2.1: Metabolic and endocrine disorders
Introduction
Pathophysiology

Clinical presentation
Imaging
CNS manifestations
MRI
Advanced neuroimaging
Hepatobiliary manifestations
Musculoskeletal manifestations
Diagnosis
Imaging differential diagnosis
Treatment and role of imaging in assessing clinical response
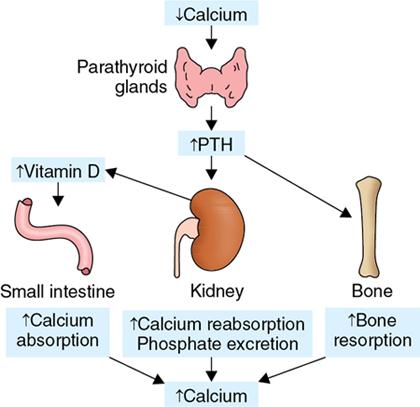
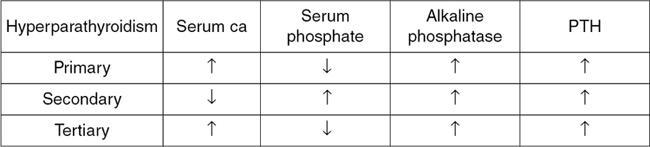
Hyperparathyroidism
Introduction
Pathophysiology (Fig. 2.1.2.1)
Clinical presentation
Serum evaluation demonstrates
Imaging
Bone resorption
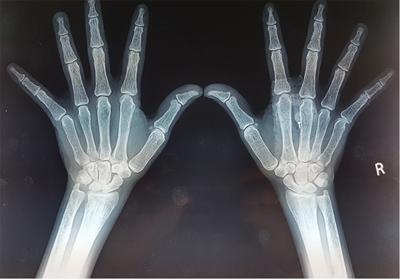
Radiographic Findings
Osteosclerosis
0
+++
Brown Tumours
+++
++
Chondrocalcinosis
++
0
Metastatic calcification
+
+++
Subperiosteal resorption
+
++
Hypoparathyroidism
Introduction
Imaging features
Radiographs and CT
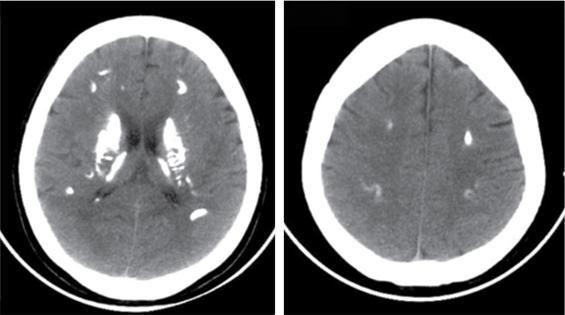
Ultrasonography
Clinical variants of PHP
Imaging manifestations
Musculoskeletal manifestations
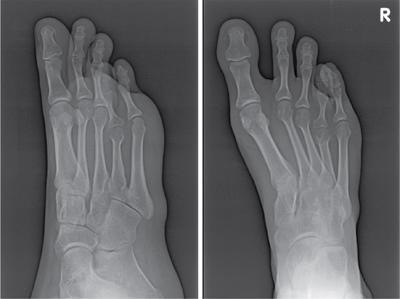
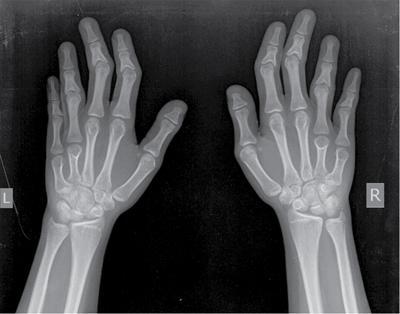
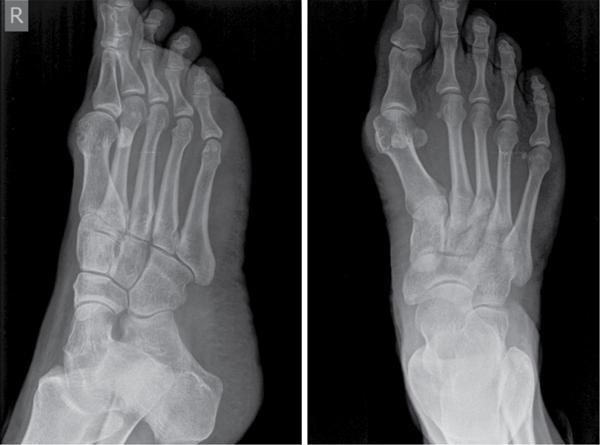
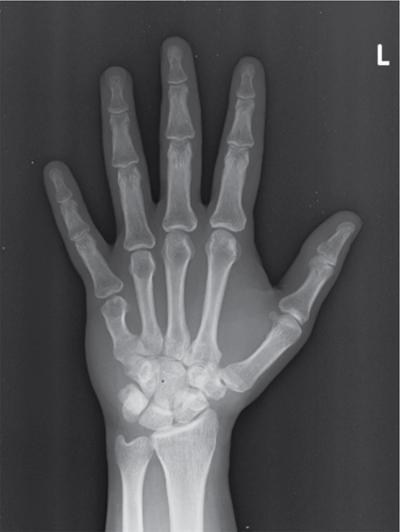
CNS manifestations
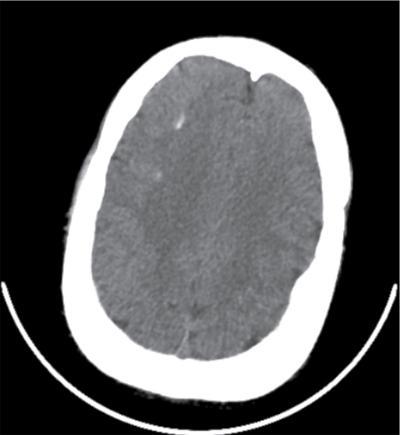
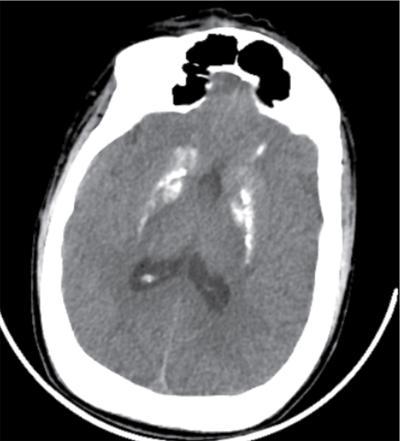
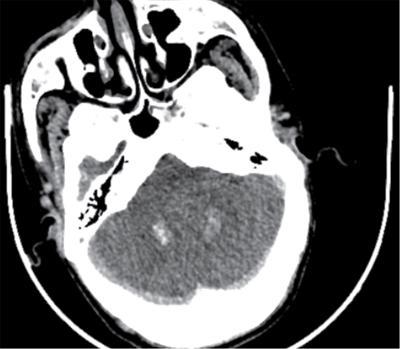
Pseudopseudohypoparathyroidism
Summary
Hyperthyroidism
Introduction
Imaging
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


