Miscellaneous Bone Diseases
INFANTILE CORTICAL HYPEROSTOSIS
Amyloidosis
Background
Amyloidosis is a rare systemic disease caused by extracellular accumulation of insoluble amyloid proteins in various organs and tissues of the body. Although several methods of classification exist, the simplest divides amyloidosis into primary and secondary forms. Amyloidosis without associated antecedent or coexisting disease is primary (idiopathic). Secondary amyloidosis is associated with chronic systemic disease (e.g., rheumatoid arthritis, Crohn disease, cystic fibrosis, chronic drug abuse), infections (e.g., familial Mediterranean fever, tuberculosis), and tumors (e.g., multiple myeloma). Usually, amyloidosis is systemic; only 10% to 20% of cases demonstrate localized disease.25,120 Primary amyloidosis affects men more than women and typically occurs between the ages of 40 and 80 years. The presentation of secondary amyloidosis depends on the associated underlying disorder.
Imaging Findings
Amyloidosis demonstrates a wide spectrum of imaging findings because a variety of systems, including the musculoskeletal, genitourinary, gastrointestinal, and cardiovascular systems, may be involved.48,116
Osteonecrosis may develop from vessel occlusion after amyloid accumulation around capillaries and endothelial cells of larger blood vessels. Other findings include osteolytic bone destruction, periarticular joint swelling, osteoporosis, pathologic vertebral fractures, joint subluxation (e.g., proximal femur and humerus), and coarse trabeculae. Calcification may be seen in the amyloid deposits.
Clinical Comments
The clinical presentation depends on the systems involved. Amyloid deposits in the heart produce pleural effusion and dyspnea. Gastrointestinal involvement is associated with bowel obstruction and malabsorption. Localized amyloid deposits in the upper respiratory system may cause hoarseness and dysphagia. Definitive diagnosis requires biopsy; imaging studies are nonspecific.
Dermatomyositis
Background
Inflammatory myopathies represent a group of disorders involving chronic inflammation, weakness, and wasting of skeletal muscle tissue. Inflammatory cells surround and destroy muscle fibers likely from an autoimmune-mediated mechanism.86 Dermatomyositis, polymyositis, juvenile myositis, and inclusion body myositis all represent inflammatory myopathies.85
Dermatomyositis represents chronic inflammation of the skeletal muscle and skin, affecting 5 of every 10,000 people. It is an easily recognized inflammatory myopathy because of the presence of a distinctive reddish rash over the eyelids, cheeks, and nose. It occurs in all ages, but is most common in adult women. Dermatomyositis is more common in children than other myopathies. In adults, it is associated with an elevated incidence of visceral carcinomas that increases with age.6,103,104
Imaging Findings
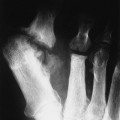
The presence of subcutaneous calcifications is the most striking radiographic feature of dermatomyositis. Subcutaneous calcifications appear commonly as linear or curvilinear radiodensities around the knees, elbows, and fingers (Fig. 15-1). Widespread calcifications (calcinosis universalis) develop in a few cases, severely limiting mobility. Calcification of intramuscular septa occurs in the deep muscles of the proximal limbs.
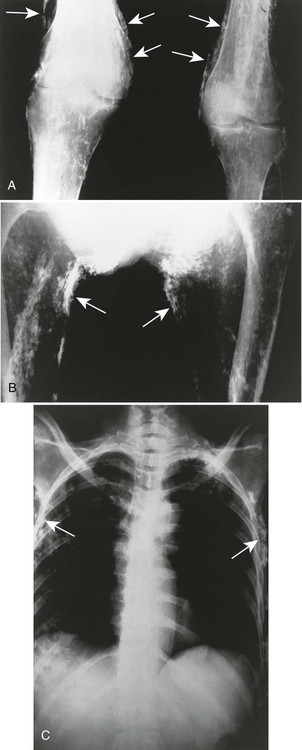
Dermatomyositis is associated with arthritis primarily in the small joints of the hands, appearing with misalignment and juxtaarticular osteoporosis and soft-tissue swelling. Characteristic is the “floppy thumb” sign, indicating subluxation of the interphalangeal joint of the first digit. A diffuse interstitial pattern is often seen on the chest radiograph.
Magnetic resonance imaging (MRI) is helpful in localizing focal inflammatory myopathy, muscle atrophy, and fatty replacement of muscle.87 MRI signal intensity correlates to the activity and distribution of the disease processes.
Clinical Comments
Dermatomyositis is not immediately painful, although it may become noticeable after muscle weakness and atrophy occur. The weakness may interfere with basic movements such as walking and raising arms. The diagnosis is made after an analysis of blood enzyme levels, electromyography, and muscle biopsy.
Steroids (e.g., prednisone) and immunosuppressive drugs (e.g., azathioprine) are given in an attempt to limit inflammation.41 Passive range of motion and ice are indicated during acute exacerbations. Moderate exercise is advocated beyond the initial inflammatory stages. Muscle stretching appears to limit limb contracture.
The course of the disease can be extremely variable, with some patients rapidly progressing to muscle wasting and weakness in just days, whereas others take years to reach this stage. Disease complications include acute renal failure and malignancy.
Gaucher Disease
Background
Gaucher disease is a lipid storage disorder resulting from a genetic deficiency of the enzyme glucocerebrosidase (glucosylceramidase), resulting in glucocerebroside accumulations within the cells of the reticuloendothelial system.78 Gaucher disease is the most common hereditary metabolic storage disorder.15
The disease may develop in individuals of any age, but is more severe in children, especially infants. A higher incidence of the disease is seen in individuals with Ashkenazi ancestry.15
Imaging Findings
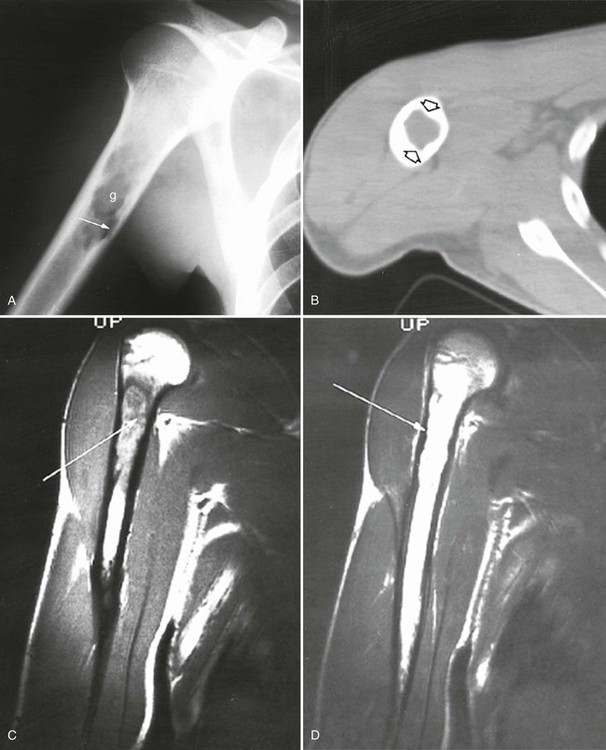
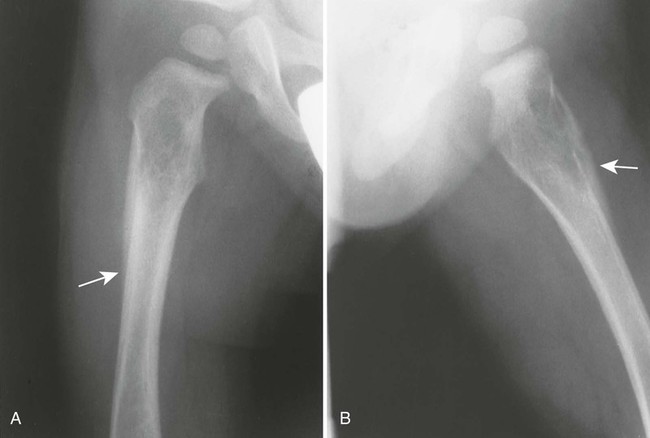
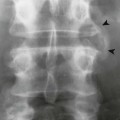
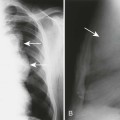
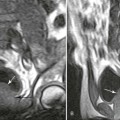
Accumulations of glucocerebroside may suppress bone marrow activity and cause destructive bone lesions and enlargement of the liver, spleen, and lymph nodes. The distal femur may exhibit thinned cortex and bone expansion (“Erlenmeyer flask” deformity). Single or multiple osteolytic lesions may occur and mimic the presentation of an infection or neoplasm (Fig. 15-2). Osteonecrosis may result from vascular occlusion, which follows the increase in the reticulum component of bone marrow (Fig. 15-3). The femoral head, humeral head, and wrist are particularly susceptible. Similar changes may produce H-shaped vertebrae at multiple levels (Fig. 15-4). MRI is useful to assess the extent and activity of bone marrow involvement (Fig. 15-5).10,55
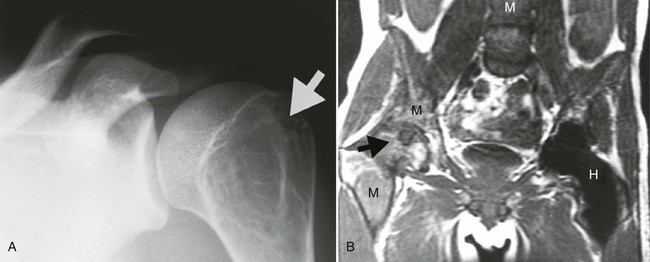
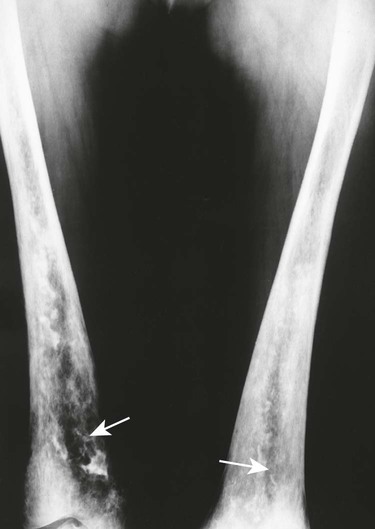
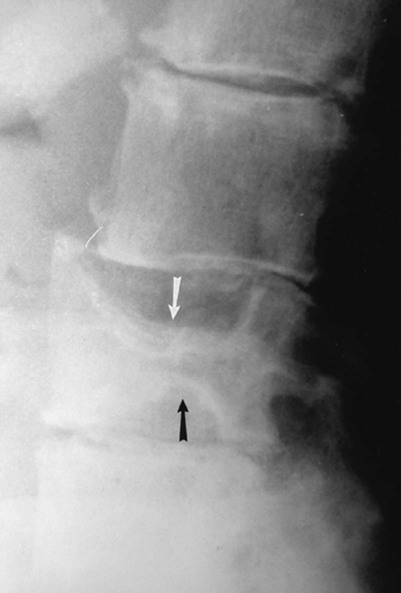
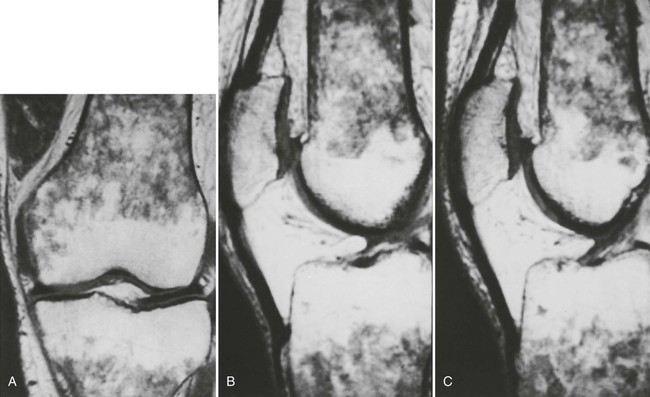
Clinical Comments
The clinical presentation of Gaucher disease is divided into three forms, based on phenotype.15 All three types are marked by hepatosplenomegaly and the presence of Gaucher cells in the bone marrow. Bone changes are more apparent in the chronic forms of the disease. The disease is more severe in infants because of cerebroside accumulations in neurons. All patients are predisposed to osteomyelitis. Intravenous infusion of glucocerebrosidase is an effective therapy but its application is limited because of cost.81
Heavy Metal Poisoning
Background
Poisoning may develop from the injection, implantation, ingestion, or inhalation of aluminum, bismuth, copper, lead, arsenic, phosphorus, mercury, or zinc. Lead is the most prevalent heavy metal, sometimes called the “silent epidemic.” Lead accumulates in the body over time, affecting multiple systems but principally the brain. Exposure is highest among occupations associated with extracting and processing lead, and among children who live in dilapidated housing whose exposure is related to the ingestion of peeling paint (Fig. 15-6). Lead-contaminated dust and soil are other potential threats of exposure for children.74
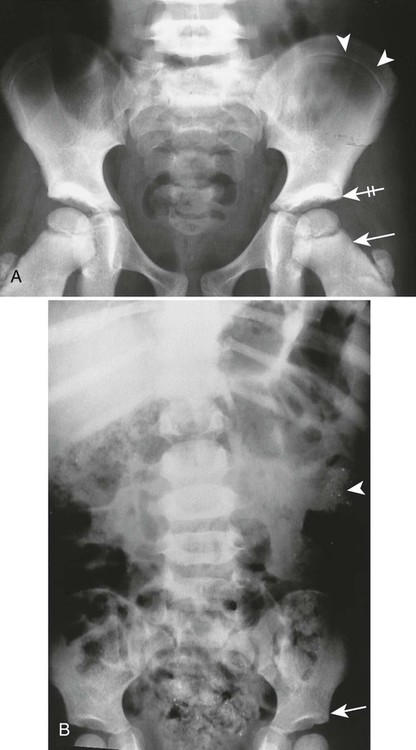
Imaging Findings
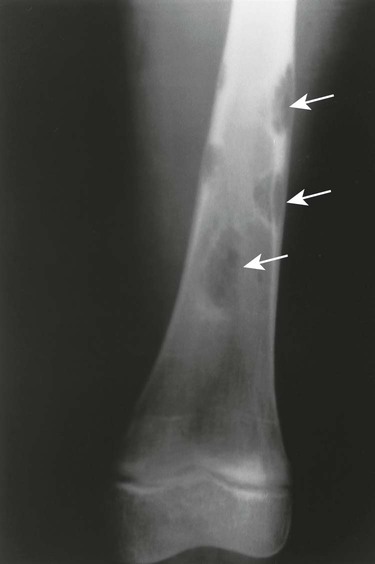
The most striking radiographic feature of lead, phosphorus, copper, or bismuth heavy metal poisoning is the presence of transverse radiodense lines in the metaphyses of long bones, especially around the knee.11,40 The density of the bands is similar to that of cortical bone. Similar lines are noted with treated rickets, scurvy, and congenital syphilis, but they may also represent a normal variant. Copper, zinc, and aluminum toxicity are associated with altered bone mineralization and osteopenia.
Clinical Comments
Heavy metal poisoning is associated with a wide variety of nonspecific clinical findings, including anorexia, irritability, apathy, abdominal colic, vomiting, diarrhea, headaches, and convulsions. Toxic exposures damage the central nervous system, blood-forming organs, gastrointestinal tract, and other systems.96,100
Histiocytosis
Background
Langerhans cell histiocytosis, formerly known as “histiocytosis X,” is the abnormal proliferation of histiocytes resulting in focal or systemic manifestations. Although the true etiology is not known, the prevailing opinion is that Langerhans histiocytosis represents a reactive rather than a neoplastic process.23 Recently, a better understanding of the disease has arisen from advances in specialized imaging and the development of immunohistochemical, morphologic, and clinical standards of diagnosis.38
Langerhans cell histiocytosis describes three clinical syndromes: eosinophilic granuloma (60% to 80% of cases), Hand-Schüller-Christian disease (15% to 40% of cases), and Letterer-Siwe disease (10% of cases). Eosinophilic granuloma is the least aggressive form, usually seen in patients between the ages of 5 and 15 years.
Hand-Schüller-Christian disease is the chronic disseminated variety, characterized by multifocal bone lesions and extraskeletal involvement of the reticuloendothelial system. Typically, it is seen in children ages 1 to 5 years. Less commonly (in 10% of cases), a clinical triad of exophthalmus, diabetes insipidus, and lytic skull lesions is present.
Letterer-Siwe disease represents an acute, disseminated, fulminant variety of the disease, seen in children younger than 2 years of age. Its aggressive clinical presentation may mimic leukemia. Most cases are fatal.
Imaging Findings
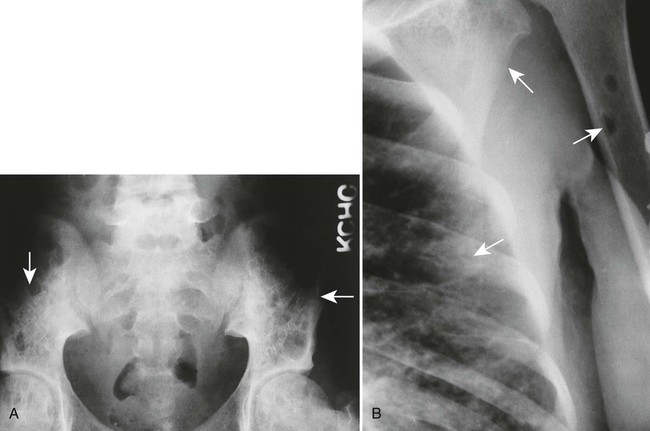
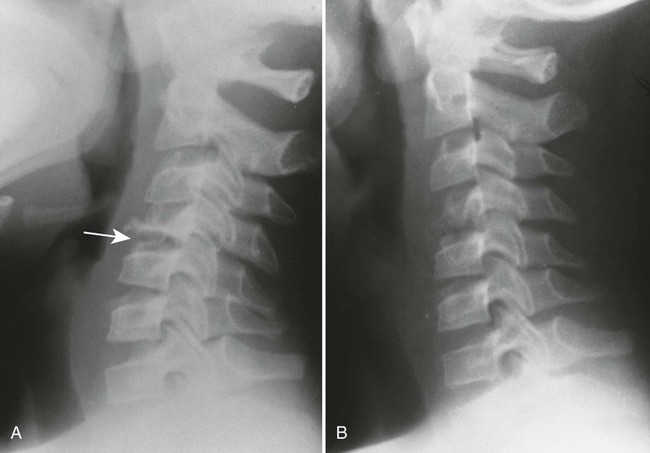
The osseous lesions of eosinophilic granuloma, Hand-Schüller-Christian, and Letterer-Siwe diseases are similar. They appear as one or more medullary-based, lytic lesions with geographic destruction (Figs. 15-7 and 15-8), lobular contour, endosteal scalloping (Fig. 15-9), periosteal reaction (long bone lesions only) (Fig. 15-10), and well-defined, uneven, or beveled margins.42 Matrix calcification and subarticular extension are not characteristic.42 Alternatively, sclerotic lesions with or without periosteal reactions may occur.
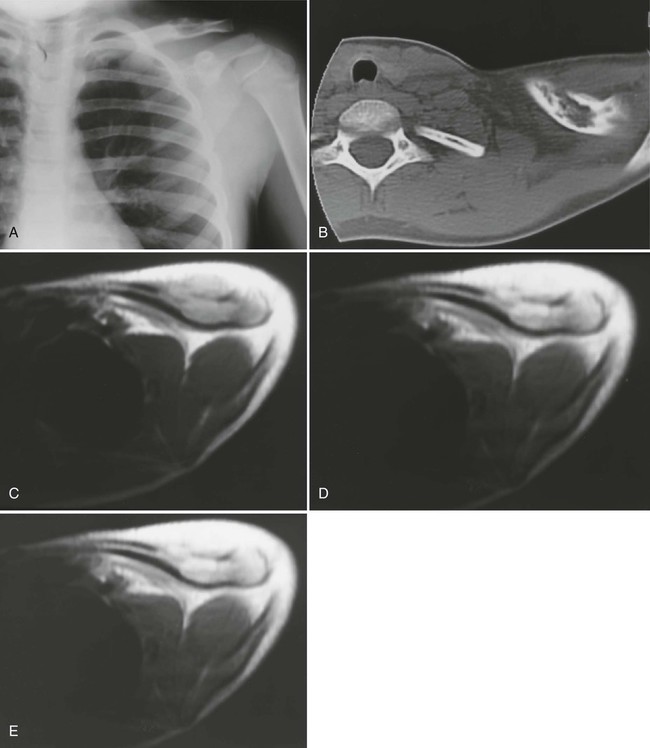
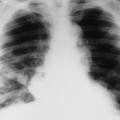
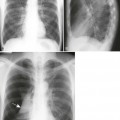
Overall, more than 50% of osseous lesions occur in the flat bones of the skull, pelvis, and ribs; about 30% of lesions occur in long bones (Fig. 15-11).114 Spinal involvement usually spares the vertebral arch and may lead to advanced vertebral collapse (vertebral plana). As the lesions heal, the diminished vertebral height may reconstitute (Fig. 15-12). Soft-tissue masses may occur, representing extension from adjacent bone marrow involvement.58 Advanced bony destruction of the mandible produces an isolated, “floating” appearance of the teeth. Pulmonary manifestation of eosinophilic granuloma includes a reticulonodular pattern of the middle and upper lung zones, often progressing to honeycomb lung.
Osseous lesions occurring with Letterer-Siwe disease typically are limited to the skull, appearing as widespread and multiple osteolytic lesions. Eosinophilic granuloma typically presents as a solitary lesion and involves the appendicular skeleton more often than Letterer-Siwe or Hand-Schüller-Christian disease. Each of the three types, especially Letterer-Siwe, may appear permeative with widespread bone destruction, mimicking findings of leukemia, Ewing sarcoma, or infection. In general, Langerhans histiocytosis has such a variable appearance that it should be considered in every destructive bone lesion that appears in patients younger than 30 years of age.
Although radionuclide scintigraphy is more sensitive than radiographic skeletal surveys in detecting histiocytic lesions in the spine, pelvis, and ribs, it is less sensitive in identifying lesions in the skull.35
Clinical Comments
Pain, fever, elevated erythrocyte sedimentation rate, progressive anemia, hepatosplenomegaly, lymphadenopathy, and diabetes insipidus may be present.33 The clinical presentation may mimic an infection. Overall, the prognosis of Langerhans cell histiocytosis is excellent in children with either localized or multifocal disease occurring in the absence of organ dysfunction, chronic disease, new-onset pituitary involvement, or long-term pulmonary fibrosis.1,76 Current therapeutic approaches involve chemotherapies, immunosuppressive drugs, bone marrow transplant, and gene therapy.1
Hypertrophic Osteoarthropathy
Background
Hypertrophic osteoarthropathy describes a primary (pachydermoperiostosis or Touraine-Solente-Golé syndrome) or secondary (Pierre-Marie-Bamberger syndrome) disorder accompanied by digital clubbing, painful swollen joints, and a symmetric, undulated, periosteal reaction.9,23,72 All features of the disorder may not be present. Although the etiology remains elusive, vascular flow, vascular endothelium, and platelet-derived growth factors have all been implicated.32,63,71 The primary form is less common (occurring in 3% to 5% of cases), has an adolescent onset and a predominance for males and blacks, and is associated with a thickened appearance of the skin of the face and scalp.
Secondary hypertrophic osteoarthropathy is associated with bronchogenic carcinoma (which occurs in up to 12% of cases),125 pulmonary abscess, pulmonary metastasis, Hodgkin disease,89 emphysema, cystic fibrosis, heart disease, and occasionally in other acute and chronic disorders.21,111 Lesions of the abdominal cavity (e.g., dysentery, Crohn disease, biliary atresia) may produce secondary hypertrophic osteoarthropathy; however, intrathoracic causes predominate, especially bronchogenic carcinoma. The age of onset is related to the underlying pathology.
Imaging Findings
The radiographic features of primary and secondary hypertropic osteoarthropathy are similar. Both are marked by periosteal reaction occurring most commonly in the tubular bones of the extremities, especially the tibia, fibula, radius, and ulna. Less commonly, the wrist, ankle, and small bones of the hands and feet are involved. The periosteal reaction is nonaggressive, thick, and widespread, occurring in the diaphysis and metaphysis (Fig. 15-13). The thickness of the periosteal reaction may increase proportionally to the duration of the disease.
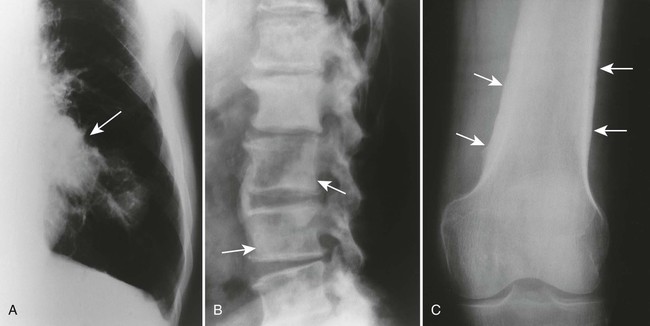
The differences between the primary and secondary forms are that the periosteal reaction occurring in patients with primary hypertrophic osteoarthropathy involves the epiphyses (secondary does not) and appears more “fluffy,” “shaggy,” or less-well-defined than does the reaction associated with secondary osteoarthropathy. Ligamentous calcifications and bony excrescences are features of primary hypertrophic osteoarthropathy, often involving the calcaneus, patella, and interosseous membrane between the radius and ulna.
Traditionally, the diagnosis of hypertrophic osteoarthropathy has been made on plain film radiographs. However, radionuclide bone scintigraphy provides a sensitive method of detection that correlates well to the clinical presentation.12,26
Clinical Comments
The clinical onset of primary hypertrophic osteoarthropathy is insidious, marked by clubbing of the distal hands and feet. The skin of the face and scalp appears thickened (pachydermia). The clinical presentation of secondary osteoarthropathy is similar but also is dependent on the underlying condition. A clinical presentation of vague bone pain and joint swelling occurs more commonly in the secondary form of the disease. The primary form usually is self-limiting after many years of involvement.
Infantile Cortical Hyperostosis
Background
Infantile cortical hyperostosis (Caffey disease, Caffey-Silverman syndrome) is an uncommon familial or sporadic syndrome marked by subperiosteal bone formation. The etiology is unknown, and it usually manifests before 6 months of age, with equal incidence in males and females.7,45,62
Imaging Findings
The radiographic features include a symmetric, thick periosteal reaction most commonly involving the mandible (80% of cases), clavicle, ulna, and less commonly the ribs, scapulae, calvaria, or diaphysis of tubular bones (Fig. 15-14).45 Although the mandible is the most commonly affected bone in the sporadic form, the tibia is the predominant bone affected in patients with the familial form.14
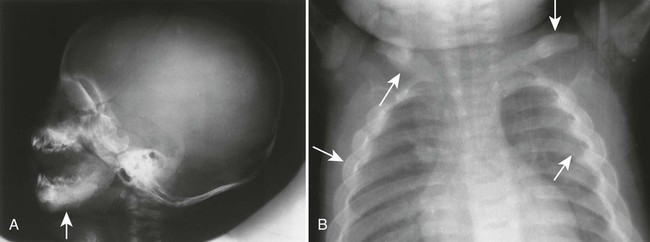
Physiologic periostitis is a common cause of periosteal reaction in infants younger than 6 months of age. A similar appearance may occur in rickets and scurvy but rarely before 6 months of age, and each demonstrates additional metaphyseal findings. Pleural effusion may accompany rib involvement. Radionuclide scintigraphy may be useful to further delineate the extent of skeletal involvement. MRI is useful to assess the presence and extent of subperiosteal hemorrhage.102
Clinical Comments
The clinical manifestation is marked by fever, hyperirritability, soft-tissue swelling over the involved bone, and often an elevated erythrocyte sedimentation rate. On palpation, the masses of involved bone and soft-tissue swelling are hard and painful. The clinical course is typically self-limiting within a few months.14
Mastocytosis
Background
Mastocytosis is a term used collectively to describe a heterogeneous group of disorders all characterized by an abnormal proliferation and accumulation of mast cells in various tissues and organs.112,121 The etiology is largely unknown, but limited evidence points to an abnormality of stem cell factor receptors.44 Mastocytosis usually is limited to the skin (90% of cases), but in rare cases it may become systemic, involving the bone marrow and gastrointestinal tract.30,44,49 Systemic disease usually manifests in adults, although a pediatric form of systemic disease exists.64 Men and women are affected equally.
Imaging Findings
The radiographic appearance of systemic mastocytosis is variable. Histamine and heparin released from the mast cells may produce generalized osteopenia similar to osteoporosis.28,59 Localized osteopenic or osteolytic defects are observed less commonly. The osteolytic lesions occur most often in the ribs, skull, and long tubular bones. Mast cell proliferation and infiltration may cause reactive sclerosis of the host bone, producing multiple osteosclerotic foci. The osteosclerotic lesions are more common in the axial skeleton and may be accompanied by thickened trabeculae. The osteolytic and osteosclerotic regions often coexist. Radionuclide bone scintigraphy may be helpful to determine the full extent of skeletal involvement.
Clinical Comments
Systemic mastocytosis comprises a wide spectrum of clinical features, depending on the organs involved, age of onset, and associated hematologic diseases.44 Clinical features are related to the release of mast cell-derived mediators (e.g., heparin, histamine, platelet-activating factor, prostaglandin, peptide leukotrienes) and include vomiting, flushing, diarrhea, hepatosplenomegaly, weight loss, and skin lesions.91 Treatment is directed toward symptom relief and the prognosis is dependent on the extent of involvement.
Neurofibromatosis
Background
Neurofibromatosis is the most common of a heterogeneous group of diseases known as phakomatoses. Phakomatoses are disorders of embryologic neuroectoderm tissue derivatives and are characterized by hamartomas of various tissues. Other phakomatoses include Lindau disease, Sturge-Weber syndrome, and tuberous sclerosis.
Neurofibromatosis is a genetic disorder affecting primarily the cell growth of neural tissue.80 At least eight presentations of the disease exist; however, only two are widely recognized: neurofibromatosis type I (von Recklinghausen disease, peripheral neurofibromatosis, or NF-1) and neurofibromatosis type II (central neurofibromatosis, or NF-2).109 Both types have an autosomal dominant pattern of inheritance, no sexual or racial predilection, and are marked by nerve sheath tumors. Each appears to be influenced by hormones, and women may notice exacerbations during pregnancy. Other than these common points, the two types appear different clinically and reflect defects of two different genes (chromosome 17 in NF-1 and chromosome 22 in NF-2).
Neurofibromatosis Type I.
NF-1 is the more common form of the disorder, affecting approximately 1 in 4000 individuals.80 It is diagnosed when at least two of the following criteria are present:
1. Six or more cutaneous macules (café-au-lait spots), larger than 5 mm before puberty, larger than 15 mm after puberty
2. Two or more neurofibromas
3. One or more plexiform neurofibromas
4. Axillary or inguinal flecking
5. Optic gliomas
6. Two or more iris hamartomas (Lisch nodules)
7. One or more characteristic bone lesions
8. First-degree relative (parent, child, or sibling) with the disease81
NF-1 is associated with a variety of intracranial hamartomatous and neoplastic lesions of the white matter and globus pallidus, including optic nerve and parenchymal gliomas.88 Those afflicted exhibit characteristic nonelevated brownish cutaneous hyperpigmentations, known as café-au-lait spots. The hyperpigmentations exhibit smooth margins (“coast of California”), as opposed to the jagged margins (“coast of Maine”) of similar hyperpigmentations that occur with polyostotic fibrous dysplasia. Other cutaneous lesions include the presence of multiple, widely dispersed soft nodules known as fibroma molluscum. In addition to the intracranial and cutaneous lesions, patients with NF-1 exhibit osseous defects of the skull, spine, and extremities, which are detailed under Imaging Findings below.51
Neurofibromatosis Type II.
NF-2 is a much less common form of neurofibromatosis, affecting 1 in 50,000 individuals.80 It is diagnosed by the presence of bilateral acoustic schwannomas or a unilateral acoustic schwannoma occurring in a patient who has a first-degree relative (parent, child, or sibling) with the disease.80 It is common for patients to develop unilateral acoustic schwannomas unrelated to neurofibromatosis. In addition to acoustic schwannomas, patients often develop schwannomas of other cranial nerves, and solitary or multiple meningiomas.115 In contrast to NF-1, cutaneous and osseous changes are not characteristic of NF-2.
Imaging Findings
Plain film radiography offers little in the evaluation of the intracranial manifestations of NF-1 or NF-2, although many of the skeletal changes occurring with NF-1 are clearly seen on plain film radiography. In rare cases, an enlarged internal acoustic or optic canal may be seen, but these osseous changes always are delineated better with computed tomography (CT). MRI offers superb imaging of intracranial and spinal lesions (Fig. 15-15). Patients with NF-1 often show hyperintense foci in the areas of brain involvement on T2-weighted images. The intensity of these areas varies over serial studies. Neurofibromas often demonstrate a characteristic pattern of peripheral hyperintensity and central hypointensity on T2-weighted images (target sign). The intracranial lesions of NF-2 (acoustic schwannomas and, less frequently, associated meningiomas) also are best imaged by MRI. On MRI film, schwannomas are hypointense or isointense relative to brain parenchyma on T1-weighted scans, are hyperintense on T2-weighted scans, and enhance after gadolinium.95
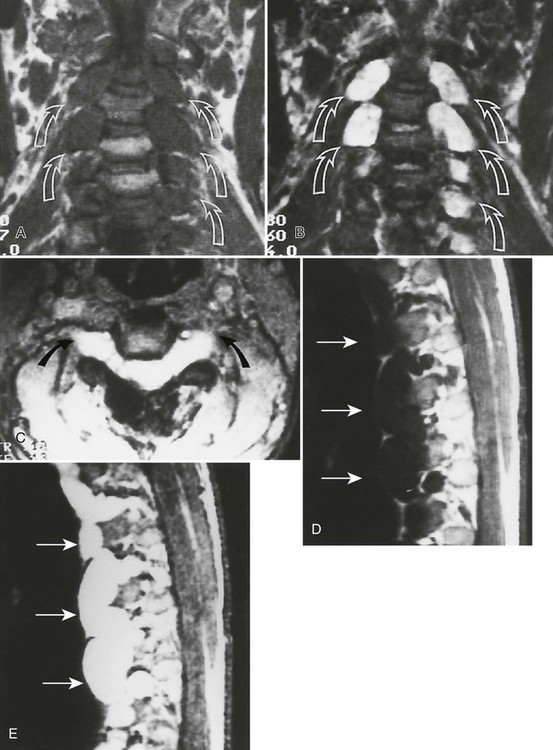
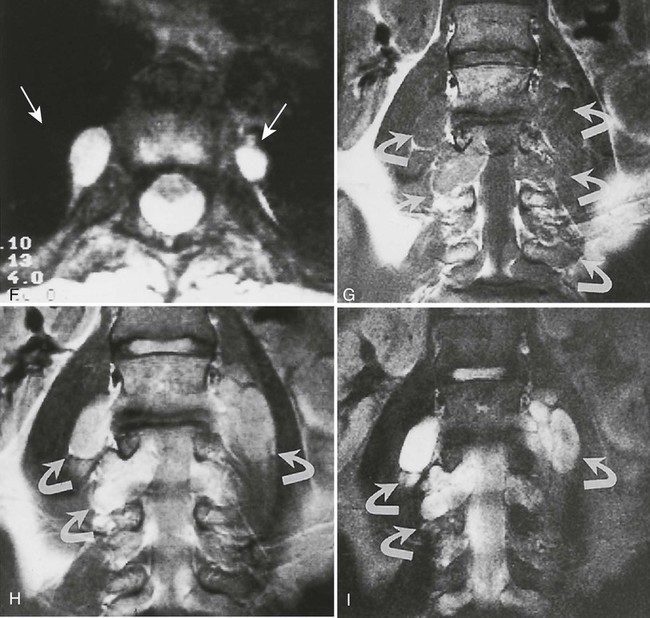
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


