Abstract
Multicystic dysplastic kidney (MCDK) denotes a kidney in which the renal parenchyma is replaced by numerous smooth-walled cysts of varying size that do not communicate with a renal pelvis and are surrounded by echogenic cortex, with an atretic ureter. It is a form of severe dysplasia that typically results in a nonfunctioning kidney. It is bilateral in approximately 25% of cases, usually conferring a lethal prognosis. Unilateral MCDK is associated with contralateral renal anomalies in 30% to 40% of cases, most commonly vesicoureteral reflux or ureteropelvic junction obstruction. Nonrenal anomalies are also frequent, occurring in approximately 25% of cases, and the finding of an associated anomaly significantly increases the fetal aneuploidy risk.
Keywords
renal dysplasia, corticomedullary differentiation, compensatory hypertrophy
Introduction
Multicystic dysplastic kidney (MCDK) is a form of severe renal dysplasia that typically results in a nonfunctioning kidney. The parenchyma is replaced by numerous cysts of varying size, with echogenic intervening tissue and an atretic ureter. Microscopically, undifferentiated epithelium and primitive ducts are surrounded by fibromuscular connective tissue. Bilateral MCDK usually results in Potter sequence (see Chapter 10 ), with early and severe oligohydramnios conferring an extremely poor prognosis secondary to pulmonary hypoplasia along with renal failure. When MCDK is unilateral, contralateral renal anomalies, in particular vesicoureteral reflux (VUR), may complicate the prognosis. The phenotypic presentation is variable, with some kidneys massively enlarged and others hypoplastic; over the course of gestation, the dysplastic kidney may enlarge, become smaller, or regress completely. Prenatal ultrasound (US) is useful for identification of this renal anomaly and, if unilateral, for surveillance of kidney size and detection of any associated abnormalities.
Disorder
Definition
MCDK is a form of severe renal dysplasia in which the kidney contains numerous smooth-walled cysts of varying size that do not communicate with a renal pelvis, surrounded by an echogenic cortex. The ureter is atretic, and the kidney is generally nonfunctioning.
Prevalence and Epidemiology
In prenatal series, the prevalence of unilateral MCDK is about 1 : 4000 births, with a reported range of 1 : 2000 to 1 : 7000. The variation likely reflects the difficulty identifying dysplastic kidneys that are small or contain few cysts. Bilateral MCDK accounts for 25% of cases, a prevalence of about 1 : 12,000 births. There is a slight male predominance for unilateral MCDK—60% of cases occur in males. When the disease is unilateral, the left kidney is more commonly affected. Bilateral MCDK is more common in females.
Unilateral MCDK is associated with contralateral renal anomalies in 30% to 40% of cases, which may have important prognostic implications because the contralateral kidney is often the only functioning kidney. The most common contralateral renal abnormality is VUR in 20% of cases, followed by ureteropelvic junction (UPJ) obstruction in 12% of cases (see Chapter 12 ). Nonrenal anomalies have also been reported in 25% of cases, including cardiac anomalies (particularly ventricular septal defects), central nervous system anomalies, spinal malformations, and gastrointestinal obstructions. Cystic dysplasia may also occur as a component of numerous syndromes, including Meckel-Gruber syndrome and Kallmann syndrome. When MCDK is found in the setting of other abnormalities, the aneuploidy risk is as high as 25%. As an isolated anomaly, unilateral MCDK is not strongly associated with aneuploidy; however, recent series have described an increased risk for pathogenic copy number variants using chromosomal microarray analysis.
Etiology and Pathophysiology
MCDK is a severe form of renal dysplasia , which comprises of a group of disorders in which nephrons and collecting ducts do not form properly, with undifferentiated epithelium, and primitive ducts surrounded by fibromuscular connective tissue. Potter proposed that MCDK was the result of a primary defect in the ureteric bud and that failure of coordination between the ureteric bud and metanephric mesenchyme resulted in abnormal development of collecting duct, loss of functional nephrons, and cyst formation. However, pathologic evaluation has shown that some fetal kidneys contain normal-appearing nephrons along with cystic dysplastic tissue, and it has been proposed that MCDK may result from impairment of urine flow early in development. This impairment may account for the association between dysplasia and contralateral renal anomalies, such as VUR.
Most cases of MCDK are sporadic, but a familial link has been reported. Ten percent of affected individuals have a first-degree relative with a renal or urinary tract malformation, or both, similar to the association found in renal agenesis. Several genes have been identified that may have important roles in the development of MCDK, including PAX2, TCF2, and uroplakins.
Manifestations of Disease
Clinical Presentation
Prenatal diagnosis is based on US findings, as discussed subsequently. In the absence of US evaluation, bilateral MCDK may manifest with lagging fundal height secondary to oligohydramnios. Historically, unilateral MCDK was a common cause of a flank mass in an infant, and diagnosis was not made in some cases until adulthood.
Imaging Technique and Findings
Ultrasound.
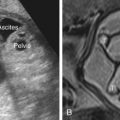
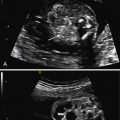
The characteristic US appearance is an enlarged kidney (or kidneys) filled with smooth-walled cysts of varying size. The cysts do not communicate, there is no normal renal pelvis, and the stroma is abnormally echogenic. In some cases, large cysts fill the cortex, and the kidney may be markedly enlarged ( Fig. 15.1 ). In others, small and medium-sized cysts are interspersed throughout the echogenic cortex ( Figs. 15.2 and 15.3 ). Rarely, MCDK may present as a single large cyst. Although the kidney may be markedly enlarged, it may also be hypoplastic and difficult to visualize, and its size may increase or decrease over time. In prenatal series of unilateral MCDK, approximately 5% of cases had completely involuted by the time of postnatal US examination. Postnatally, more than half will demonstrate partial or complete regression over time.