Multisystem Disorders with Pulmonary Manifestations
Teresa Chapman, MD
LEARNING OBJECTIVES
1. Recognize distinctive radiographic features of cystic fibrosis (CF), and differentiate CF from immotile cilia syndrome based on distribution of lung disease.
2. List additional extrathoracic systems impacted by CF.
3. Define acute chest syndrome (ACS), and indicate differences in the clinical presentation of children versus adolescents and young adults.
4. Discuss the cause of right heart enlargement in sickle cell disease (SCD) patients.
5. Recognize typical high-resolution chest CT patterns of pulmonary involvement by Langerhans cell histiocytosis.
6. Identify high-resolution chest CT findings in pediatric patients with mixed connective tissue diseases, dermatomyositis, lupus, and juvenile idiopathic arthritis.
7. Recognize atypical thoracic presentations of systemic lupus erythematosus.
INTRODUCTION
Multisystem diseases that involve the chest in the pediatric population may rely on diagnostic imaging for diagnosis, or may simply require imaging for surveillance of disease or response to therapy. In this chapter, we focus on a small number of disease entities that are either common, and therefore essential to know well, or unusual but offer the reader a fundamental understanding of how to approach nonspecific findings in the chest. Chest radiography is frequently the initial imaging study for systemic diseases manifesting with vague symptoms. The radiologist must be comfortable considering the potential implications of various pulmonary parenchymal patterns of disease, which are discussed here in the context of specific diagnoses. As always, diagnostic clues beyond the lungs must also be observed. This chapter will discuss the following systemic diseases seen in children: CF, SCD, Langerhans cell histiocytosis (LCH), and systemic autoimmune disorders.
CYSTIC FIBROSIS
CF is one of the most common severe autosomal recessive disorders in the Caucasian population, with an incidence of 1 in 3,000 live births.1 The gene responsible for this disease is called the CF transmembrane conductance regulator (cftr) gene, located on chromosome 7, and it encodes a large protein that functions as a cyclic AMP-dependent chloride channel.2,3 This transmembrane channel normally localizes to the membranes of secretory and absorptive epithelial cells within the airways, pancreas, liver, intestine, sweat glands, and vas deferens. The effect of a mutation in the cftr gene is insufficient hydration of large molecules within duct lumens in these organs. Damage arises from ductal or glandular obstruction by the concentrated secretions in these multiple organs. A physician suspecting the disease may pursue “sweat testing,” a laboratory diagnostic test that involves the quantitative or qualitative analysis of sweat to determine chloride concentration for confirmation of a CF diagnosis; a level of 60 mEq/L or greater is diagnostic of CF.4 Over 1,950 mutations of the cftr gene have now been described,5 and the phenotypic manifestations of any given individual vary widely. Therapies in development are directed at modifying the CF transmembrane conductance regulator, depending on an individual’s specific membrane transporter defect.6
Pulmonary manifestations of CF are the main cause of morbidity in these patients, and children with CF are followed routinely with pulmonary function tests and basic spirometry.7,8 Data from a multicenter trial suggested that spirometry outcomes in preschool patients with CF are more discriminative than are those from more elegant and involved pulmonary function tests, such as forced oscillation or inductance plethysmography.8 In infants and toddlers, however, lung disease has been shown to be evident by CT imaging of the chest, even prior to detecting abnormalities on pulmonary functions tests.9 Quantification of lung disease with validated CT scoring systems allows for longitudinal monitoring of observed changes.10, 11 and 12 However, concerns regarding exposure to ionizing radiation continue, even in the setting of improving dose-reduction techniques. Interestingly, validated radiographic scoring systems have been shown to be equally reliable to CT scoring systems.13, 14 and 15 Important radiographic features in these scoring systems include air trapping, linear markings (reflecting bronchial
wall thickening), nodular cystic lesions (reflecting bronchiectasis), large lesions such as atelectasis and pneumonia, and general severity of disease.13,15
wall thickening), nodular cystic lesions (reflecting bronchiectasis), large lesions such as atelectasis and pneumonia, and general severity of disease.13,15
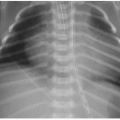
Early evidence of pulmonary involvement is air trapping from small airways obstruction, which is detectable on expiratory radiographic views of the chest (Fig. 8.1). Persistent or recurrent presentations of viral-like illness, or persistent peribronchial thickening on chest radiography following a previous episode of presumptive viral infection or reactive airways disease, should prompt consideration of CF as an underlying cause. Bronchiectasis is an inevitable finding and, as mentioned above, may be present even in the setting of normal lung function testing.10,16,17 Bronchiectasis and peribronchial thickening develop predominantly in the upper lobes (Fig. 8.2). The other main finding is mucous plugging, which manifests as nodular opacities on chest radiography and as centrilobular nodules on CT (Fig. 8.3).
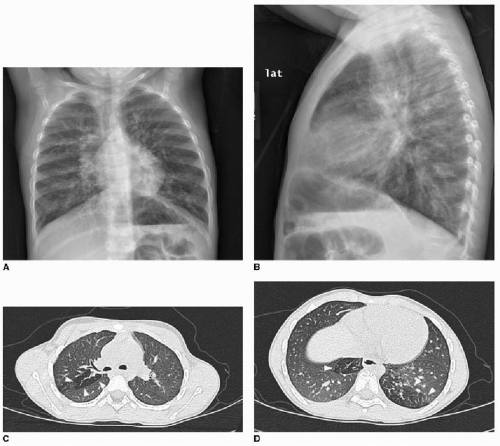 FIG. 8.1 • Air trapping as a reflection of small airways disease is cystic fibrosis. Large lung volumes on frontal (A) and lateral (B) chest radiographs in a 2-year-old male with CF show flattening of the diaphragm, indicating air trapping from the small airways inflammation. Central airways are also abnormally thickened bilaterally. In a different 4-year-old patient with milder disease, axial CT images (C,D) show areas of pulmonary parenchyma with lower attenuation (arrowheads), indicating regional small airways disease. |
A differential consideration for bronchiectasis is immotile cilia syndrome (also known as primary ciliary dyskinesia), which, unlike CF, is an uncommon autosomal recessive disease that also affects mucociliary transport. In this disease, radiographic manifestations in children include peribronchial thickening, mucous plugging, bronchiectasis, consolidations, and less commonly air trapping. The imaging finding that suggests this disease rather than CF is a mid- and lower-lung predominance rather than an upper lobe distribution.18 Kartagener syndrome refers to immotile cilia syndrome combined with situs inversus (Fig. 8.4). Pectus excavatum and polysplenia are associated with a small number of these cases.19
The respiratory tract involvement in CF also manifests in the upper airway, specifically in the paranasal sinuses with obstruction and nasal polyps.1,20 Gastrointestinal manifestations of CF are numerous and include pancreatic insufficiency, pancreatitis
(Fig. 8.5), hepatic fibrosis with portal hypertension, meconium ileus in the neonate (seen in 13% of neonates with CF) and distal intestinal obstruction syndrome later in life, gastroesophageal reflux disease (GERD), and eosinophilic esophagitis.21,22 Medical therapies for CF are directed toward avoiding the long-term effects of maldigestion and malabsorption of nutrients due to gastrointestinal disease. The vast majority of CF patients take exogenous pancreatic enzyme supplements routinely. Proton pump inhibitors are the primary line of therapy for GERD, and it has been shown that lung function is better in children with CF treated for GERD than in those who are not on acid suppressive medication.23 Apart from pulmonary and GI manifestations, the other main complication of CF to be aware of is infertility in males.
(Fig. 8.5), hepatic fibrosis with portal hypertension, meconium ileus in the neonate (seen in 13% of neonates with CF) and distal intestinal obstruction syndrome later in life, gastroesophageal reflux disease (GERD), and eosinophilic esophagitis.21,22 Medical therapies for CF are directed toward avoiding the long-term effects of maldigestion and malabsorption of nutrients due to gastrointestinal disease. The vast majority of CF patients take exogenous pancreatic enzyme supplements routinely. Proton pump inhibitors are the primary line of therapy for GERD, and it has been shown that lung function is better in children with CF treated for GERD than in those who are not on acid suppressive medication.23 Apart from pulmonary and GI manifestations, the other main complication of CF to be aware of is infertility in males.
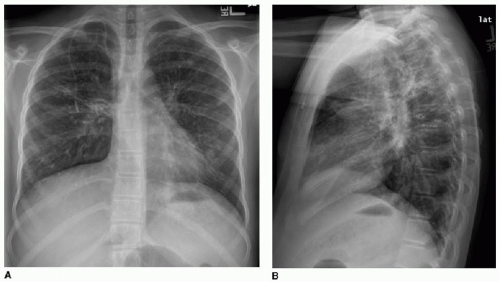 FIG. 8.2 • Predominantly upper lobe bronchiectasis seen radiographically in a 12-year-old male with cystic fibrosis. PA (A) and lateral (B) radiographs of the chest show cylindrical lucencies radiating from the superior hila, reflecting the dilated airways, much more pronounced in the upper lobes than the lower lobes. |
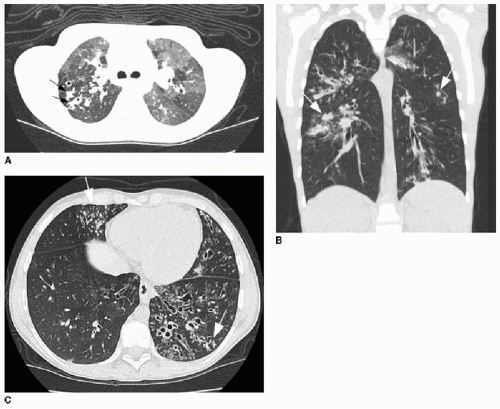 FIG. 8.3 • Nodules and bronchiectasis on chest CT in two different patients with cystic fibrosis. Axial (A) and coronal (B) images from a chest CT study in an 11-year-old female with CF show extensive bronchiectasis and nodules (arrows), typical of this disease. Note also the heterogeneity of the background lung parenchyma, reflecting the variable distribution of small airways disease. Axial high-resolution chest CT in a different patient (C), age 12 years, shows a different distribution of disease, disproportionately affecting the left lower lobe with marked bronchiectasis. Scattered small centrilobular nodules (arrows) are appreciable, reflecting mucous plugging within smaller airways. This lower lobe distribution prompts consideration of immotile cilia syndrome rather than CF, although the case shown here is an atypical example of CF. |
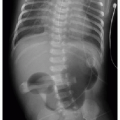
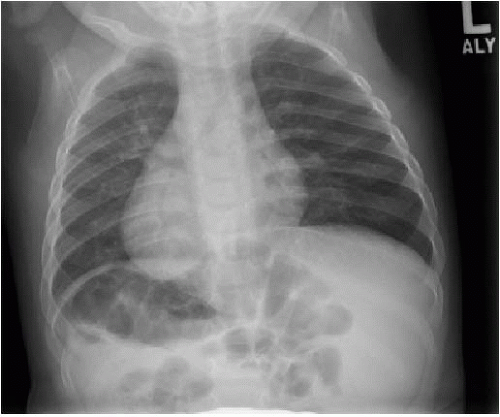 FIG. 8.4 • Kartagener syndrome in a 4-year-old child. Situs inversus is evident on this frontal chest radiograph, with the cardiac apex directed toward the right and hepatic shadow on the left. Transverse colonic gas overlaps with the gastric bubble, located on the right. |
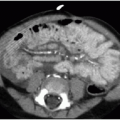
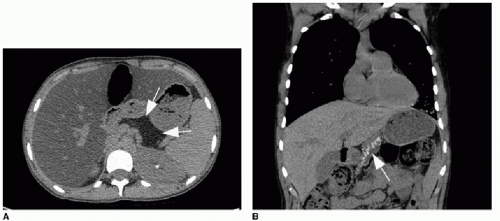 FIG. 8.5 • Abdominal findings in cystic fibrosis. Axial image through a noncontrast CT study of the abdomen (A) in a 16-year-old female demonstrates fat attenuation in the expected location of the pancreas (arrows), and the liver shows substantially diffusely lower attenuation than does the spleen. Findings are consistent with fatty replacement of the pancreas and hepatic steatosis. In a different 18-year-old patient with CF, coronal reconstruction of a noncontrast CT through the chest and abdomen (B) shows abnormal coarse calcifications through the pancreas, consistent with chronic pancreatitis. |
SICKLE CELL DISEASE
SCD is a common autosomal recessive blood disorder that affects millions of people worldwide, with an incidence of approximately 1 in every 500 African American births and 1 in every 1,000 to 1,400 Hispanic American births.24 The disease originates from a monogenetic mutation causing a single amino acid substitution in the beta chain of hemoglobin and results in distortion of the red blood cell shape under stressed conditions. This change in cellular morphology leads to hemolysis and anemia, recurrent vasoocclusive events, and ultimately end-organ failure.25
ACS is an acute lung injury defined as the presence of a new segmental pulmonary infiltrate along with fever, chest pain, respiratory distress, or new-onset hypoxemia.26 It is a frequent cause of hospitalization and is the most common cause of SCD-related death in these patients.27 Known inciting causes of ACS in sickle cell patients include fat emboli (due to a vasoocclusive crisis in multiple bones), pulmonary infarction from affected erythrocytes, asthma, and infectious etiologies such as Mycoplasma pneumoniae and Streptococcus pneumoniae.28,29 The clinical features of ACS overlap with infectious pneumonitis, making the diagnosis difficult. Children younger than 10 years of age present with cough, wheezing, and fever. Adolescents may present more like adults, with sudden onset of shortness of breath, severe pain, chills, and rapid progression to respiratory failure.26,30
Diagnostic imaging for chest symptoms in a child or adolescent with SCD utilizes chest radiography to identify any pulmonary opacification and to diagnose alternative explanations for the symptoms. As mentioned above, the main finding will be pulmonary consolidation affecting at least one segment (Fig. 8.6). Any amount of pulmonary opacification, however, is important
in a sickle cell patient, since pulmonary infection and infarction may manifest with a smaller area of pulmonary opacification and may lead to development of ACS. Asthma, which affects approximately 15% to 20% of African American children,28 is a small airways reactive disorder caused by inflammation and typically results in trapping of air distal to the obstructed small airways. The hypoxia caused by asthma can induce red blood cell sickling, and therefore ACS is seen more commonly in the setting of asthma. This is evident radiographically by large lung volumes, and manifests on chest CT imaging by a mosaic pattern of pulmonary parenchymal attenuation (reflecting contrasting regions of normal and hyperlucent lung). As SCD advances, pulmonary hypertension also can develop,28 and this may be reflected by right heart enlargement (Fig. 8.7).
in a sickle cell patient, since pulmonary infection and infarction may manifest with a smaller area of pulmonary opacification and may lead to development of ACS. Asthma, which affects approximately 15% to 20% of African American children,28 is a small airways reactive disorder caused by inflammation and typically results in trapping of air distal to the obstructed small airways. The hypoxia caused by asthma can induce red blood cell sickling, and therefore ACS is seen more commonly in the setting of asthma. This is evident radiographically by large lung volumes, and manifests on chest CT imaging by a mosaic pattern of pulmonary parenchymal attenuation (reflecting contrasting regions of normal and hyperlucent lung). As SCD advances, pulmonary hypertension also can develop,28 and this may be reflected by right heart enlargement (Fig. 8.7).
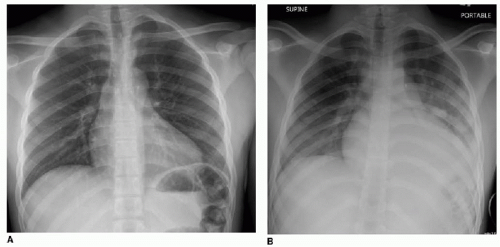 FIG. 8.6 • Acute chest syndrome in a 14-year-old child with sickle cell disease. A baseline frontal chest radiograph (A) shows clear lungs. Five months later (B), the same patient presents with chest pain and dyspnea and has left lower lobe opacification as well as hazy airspace opacities in the left upper lobe and right lower lobe. |
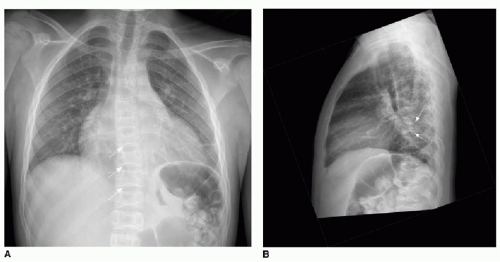 FIG. 8.7 • Nonpulmonary findings seen radiographically in a 14-year-old child with sickle cell disease. Frontal (A) and lateral (B) radiographs in this child show mild cardiomegaly, reflecting the right heart enlargement that develops secondary to pulmonary hypertension. Vertebral endplate depressions (arrows) reflect recurrent osseous microinfarctions, creating the “H-shaped” vertebrae typical of this disease. Note also the absence of a splenic shadow, which disappears due to splenic autoinfarction. |
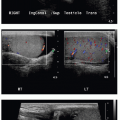
Diagnosis of SCD may be suspected on radiography or CT, even without cardiopulmonary disease, by the appearance of the vertebrae. Microinfarction of the thoracic vertebrae endplates leads to depressions that have been termed “H-shaped” or “Lincoln log” vertebrae, and this is seen in approximately 10% of patients with sickle cell anemia (Fig. 8.7).31 Autoinfarction of the spleen occurs in older children and adults; the spleen is usually larger than expected in young children with SCD and appears heterogeneous on ultrasound.31 Cholecystectomy clips in the right upper quadrant are commonly seen in SCD patients, as well, due to the high incidence of cholelithiasis.
Patients with ACS are treated with hospital admission, intravenous antibiotics, bronchodilators, oxygen supplementation, and pain management (in the adolescent). Incentive spirometry is used as a means to prevent progression.32 In severe cases, ventilation and rarely extracorporeal life support (ECLS) are required.33,34
LANGERHANS CELL HISTIOCYTOSIS
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


