the setting of a normally formed corpus callosum, and they have no clinical implication (Fig. 30.3).
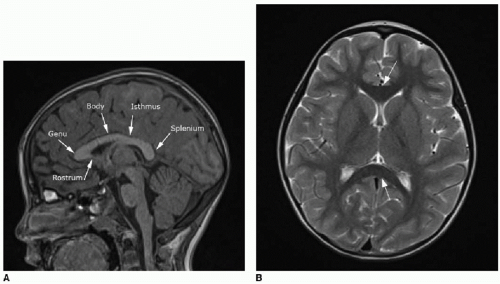 FIG. 30.1 • Corpus callosum anatomy on brain MRI of normal 5-year-old child. A: Sagittal T1-weighted MR image shows large hyperintense bundle of midline crossing myelinated fibers. Components of normal corpus callosum are annotated here. B: Axial T2-weighted MR image shows dark signal of myelinated fibers anteriorly and posteriorly (arrows). |
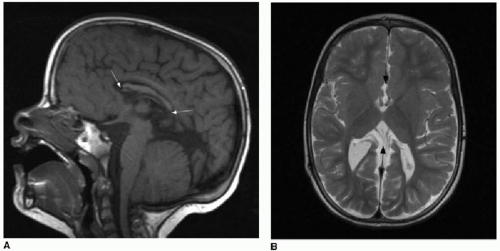 FIG. 30.2 • Corpus callosum partial dysgenesis and thinning. Sagittal T1-weighted (A) and axial T2-weighted (B) MR images from a 4-year-old child with Chiari malformation and corpus callosal dysgenesis show partial absence of corpus callosum. Posterior body, splenium, and rostrum are absent (white arrows on A). Axial image shows absence of crossing fibers (black arrows in B) that should be readily apparent. Lateral ventricles assume a parallel orientation, typical of partial and complete corpus agenesis. Sagittal T1-weighted (C) and axial T2-weighted (D) MR images from 5-month-old infant with large left hemispheric porencephalic cyst (PC) show a fully formed but markedly thinned (white arrows) corpus callosum secondary to white matter volume loss. Note normal convergence of lateral ventricles toward one another on axial plane, in contrast to partial dysgenesis case. |
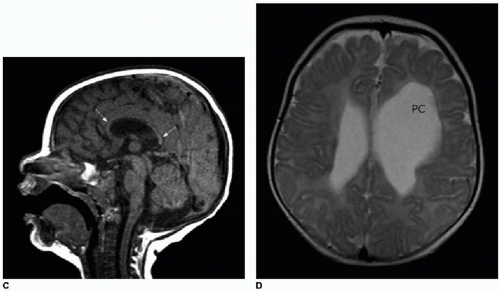 FIG. 30.2 • (Continued). |
of diffuse proliferative or migrational disorders characterized by abnormal simplification of the cortical convolutions (Fig. 30.4).10,11 Agyria refers to absence of cortical gyri, whereas pachygyria refers to abnormally broad cerebral gyri; these terms also fall within the category of lissencephaly. A number of genetic defects causing lissencephaly have been identified.11 While diffuse migrational anomalies present with severe developmental delay, intractable seizures, and hypertonia, the focal malformations tend to present with medical refractory seizures, and we will focus on these entities here.
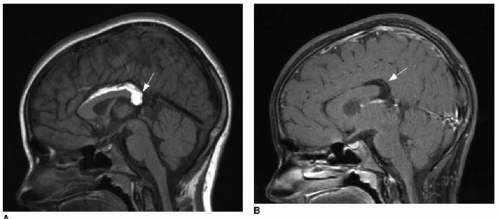 FIG. 30.3 • Interhemispheric/callosal lipoma. Sagittal T1-weighted MR image without (A) and with (B) fat saturation in a 10-year-old female shows fat signal in the midline, along the mid- and posterior corpus callosum (arrow), consistent with interhemispheric lipoma. |
Table 30.1 CATEGORIZATION OF NEURONAL MIGRATION ANOMALIES | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
addition to seizures.7 A diffuse form of gray matter heterotopia is called subcortical band heterotopia (SBH), wherein a thick band of tissue isointense to gray matter replaces a variable volume of white matter, and only a thin rim of white matter is evident between the heterotopic tissue and the cortex (Fig. 30.4). SBH falls into the category of lissencephaly, and its clinical severity depends on the extent of cortical abnormalities, band thickness, and ventricular enlargement.11
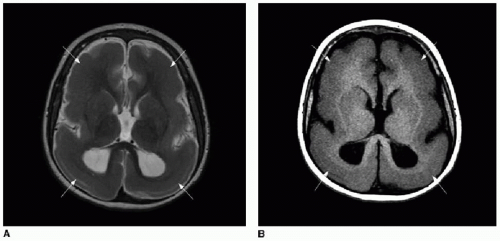 FIG. 30.4 • Lissencephaly. Axial (A) T2-weighted and T1-weighted (B) MR images of 7-year-old patient’s brain show markedly simplified cerebral sulcation pattern bilaterally. Widespread replacement of expected subcortical white matter signal is abnormally replaced by gray matter signal (arrows), consistent with diffuse subcortical band heterotopia. |
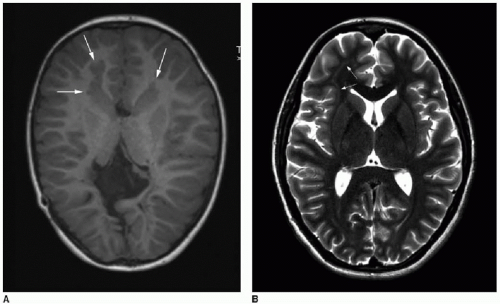 FIG. 30.5 • Gray matter heterotopia. A: Periventricular gray matter heterotopia in bifrontal lobes (arrows) shown on axial T1-weighted MR image of a 5-year-old child with seizures. Note also complete agenesis of corpus callosum. B: Subcortical thin band of heterotopia (arrows) in right frontal white matter shown on axial T2-weighted MR image of 17-year-old patient with seizures. Signal of abnormal white matter nodules and band followed gray matter signal on all sequences (not shown here). |
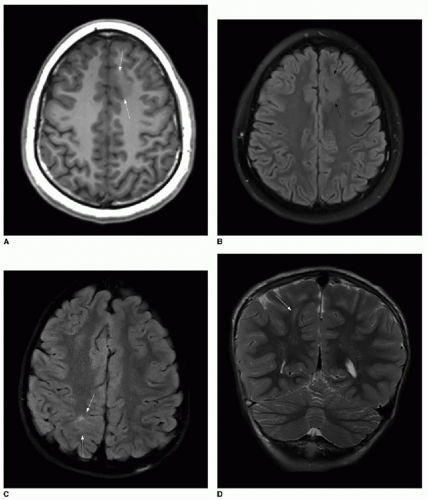 FIG. 30.6 • Focal cortical dysplasia. Type I FCD in 15-year-old patient with seizures is evident on T1-weighted (A) and T2-weighted (B) axial images as area with loss of gray-white matter differentiation and abnormal signal on both sequences (arrows). C: Type II FCD in 5-year-old patient manifests with hyperintense T2-weighted signal on fluid attenuation and inversion recovery (FLAIR) sequence in axial plane (arrows). D: Transmantle sign is apparent on coronal T2-weighted image, with high signal radiating from affected cortex toward the ventricular surface. Following surgical resection, histopathology confirmed presence of balloon cells. |
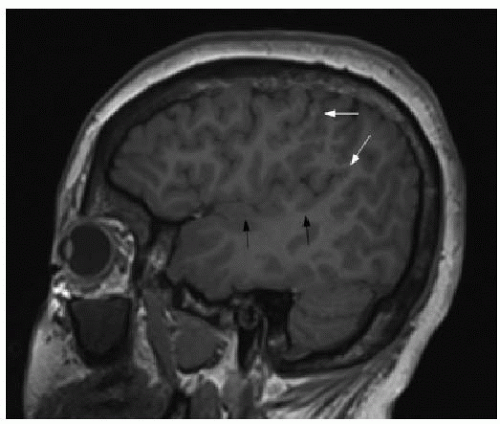 FIG. 30.7 • Perisylvian polymicrogyria. Sagittal T1-weighted thinslice MR image of 16-year-old seizure patient’s brain shows excessive number of small cortical gyri at upper aspect of perisylvian region (white arrows), consistent with PMG. Contrast with normal gyral pattern along Sylvian fissure more inferiorly (black arrows). |
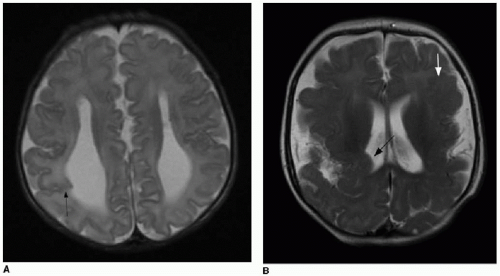 FIG. 30.8 • Examples of schizencephaly. A: Axial T2-weighted brain MR image of male neonate with prenatal diagnosis of agenesis of corpus callosum and ventriculomegaly demonstrates abnormal gray matter-lined right parietal cleft coursing from cortical surface to ventricular surface (arrow). Note parallel configuration of lateral ventricles, typical of absent corpus callosum. B: Axial T2-weighted brain MR image of 5-month-old female infant at the level of lateral ventricles shows extensive malformation of cerebral hemispheres. Right parietal cleft anomaly creates indentation upon ependymal surface (black arrow), characteristic of schizencephaly. Dysplastic gray matter lines the cleft. Note also polymicrogyria in left frontal lobe (white arrow). |
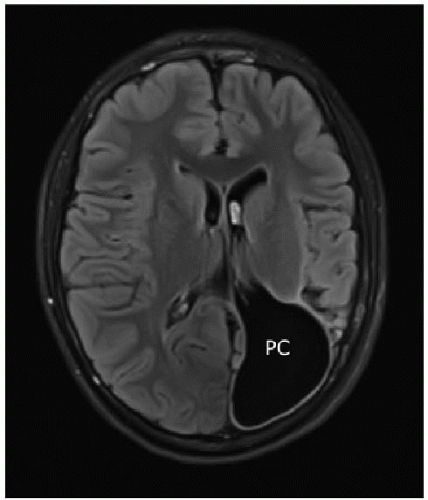 FIG. 30.9 • Porencephalic cyst. Axial T2-weighted image of the brain of 13-year-old shows large white matter-lined CSF space contiguous with left lateral ventricle. (PC, porencephalic cyst.) |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


