Solid Organs of the Gastrointestinal Tract
Teresa Chapman, MD
LEARNING OBJECTIVES
1. Differentiate neonatal hepatitis from biliary atresia using ultrasound and scintigraphy.
2. Classify various types of choledochal cysts, and discuss appropriate imaging for evaluation.
3. List intrahepatic tumors associated with elevated alpha-fetoprotein levels.
4. Indicate useful imaging features that can differentiate fibrolamellar hepatocellular carcinoma from focal nodular hyperplasia.
5. List two features that distinguish congenital hemangiomas from infantile hemangiomas.
INTRODUCTION
There is considerable overlap between pediatric and adult conditions that affect the solid organs of the gastrointestinal tract. Malignancies and inflammatory disorders may involve the liver and pancreas in the child, as in adults. In this chapter, common and significant diagnoses involving the liver are primarily discussed, with an emphasis on how the age of presentation influences the differential diagnosis. A limited discussion of pediatric pancreatic masses is provided as well.
BILIARY ATRESIA
Biliary atresia refers to a congenital progressive cholangiopathy affecting the intrahepatic and extrahepatic biliary system to variable degrees.1, 2 and 3 The resulting presentation is neonatal jaundice secondary to obstructive cholestasis. Without treatment, the disorder may progress to hepatic fibrosis, cirrhosis, liver failure, and death by age 2 years.1,2 The disease has a much higher incidence in Asian countries (1 in 5,400 to 10,000 live births) than in Western countries (1 in 15,000 to 19,000 live births),4,5 and its pathogenesis is likely multifactorial, with both genetic and environmental factors influencing its development.6 The vast majority of cases (90%) show a configuration wherein the most proximal part of the extrahepatic biliary duct in the hepatic portal region is fibrotic, and there is no macroscopic remnant of the hepatic duct. Microscopic bile ductules may, however, remain in continuity with the intrahepatic biliary tree. Cystic dilatation of the extrahepatic bile duct remnants may be found.2
It is common for the neonate to have some transient hyperbilirubinemia. This is because of physiologic breakdown of red blood cells to normalize the polycythemia that is present after birth, combined with the neonatal liver’s inability to rapidly clear bilirubin.7 Physiologic neonatal jaundice should resolve within 1 week after birth. There is an extensive list of possible causes of pathologic hyperbilirubinemia in the newborn, including hemolytic disease, birth trauma, infection, medications, and metabolic disorders. When a newborn infant with jaundice is referred for diagnostic imaging, the main differential considerations are biliary atresia and neonatal hepatitis.8 The distinction between the two diagnoses is critical, because biliary atresia must be treated surgically with a hepatoportoenterostomy (Kasai procedure), which establishes bile drainage from the microscopic residual bile ductules directly into the bowel, whereas neonatal hepatitis is treated medically.1, 2 and 3,8
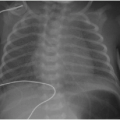
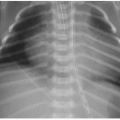
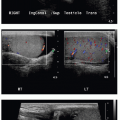
The diagnostic imaging modalities relevant to distinguishing between these two different causes of neonatal jaundice include ultrasonography, hepatobiliary scintigraphy, and magnetic resonance cholangiopancreatography (MRCP). Because of its wide availability, US continues to be a first-line imaging modality. It has been shown that sonographic diagnosis of biliary atresia is possible using three findings: the triangular cord sign (TCS),9 gallbladder length, and gallbladder contractility. Contractility may be measured by the following formula: [(fasting volume — postprandial volume) ÷ fasting volume] × 100.10 The TCS refers to an echogenic band of tissue at the porta hepatis (Fig. 16.1A), which correlates with fibrosis in this region intraoperatively,9 and is seen approximately in 80% of biliary atresia cases.10 A gallbladder length less than 15 mm (including a nonvisualized gallbladder) after fasting for more than 3 hours is seen in 77% of biliary atresia cases, compared to only 27% of nonbiliary atresia cases (Fig. 16.1B). It is important to keep in mind that a gallbladder may appear normal in approximately 20% of biliary atresia cases. Although the gallbladder contractility is more likely to be abnormally low (less than 86%) in cases of biliary atresia, this finding has a lower specificity and (72%) and positive predictive value (67%) than do the findings of TCS (85%/95%) and small gallbladder (77%/79%)10; furthermore, time restrictions may make this assessment impractical. Doppler ultrasound contributes further to the diagnosis of biliary atresia by recognition of two features: a hepatic artery diameter greater than 2 mm is more commonly
seen with biliary atresia (2.5 ± 0.55 mm) than nonbiliary atresia (1.9 ± 0.6 mm),11,12 and the presence of subcapsular vessels (Fig. 16.2) is almost exclusively seen in biliary atresia cases.12
seen with biliary atresia (2.5 ± 0.55 mm) than nonbiliary atresia (1.9 ± 0.6 mm),11,12 and the presence of subcapsular vessels (Fig. 16.2) is almost exclusively seen in biliary atresia cases.12
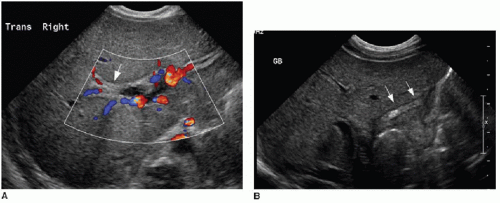 FIG. 16.1 • Sonographic findings of biliary atresia in two separate infants. A: Two-day-old infant with conjugated hyperbilirubinemia and ultrasound notable for triangular cord sign (arrow), reflecting fibrotic tissue along the secondary biliary duct branches. B: Three-month-old infant with biliary atresia and small, contracted gallbladder (arrows). |
Scintigraphic evaluation of the neonatal liver and biliary tree (HIDA scan) is performed using either technetium-99m mebrofenin or 99m-Tc-disofenin, which are taken up by hepatocytes and excreted into the biliary system.13 A common protocol relies on the administration of phenobarbital or ursodeoxycholic acid for 5 days prior to the study, to stimulate satisfactory hepatocyte activity.14,15 The radiotracer (0.05 mCi/kg) is injected intravenously, and anterior planar imaging of the abdomen is performed at short intervals over 60 minutes, followed by delayed imaging at 6 and 24 hours. Normal hepatobiliary findings include rapid clearance of blood pool activity and immediate demonstration of hepatic parenchyma, followed sequentially by radiotracer visualization in the intra- and extrahepatic biliary ductal tree, gallbladder, and upper small bowel within 1 hour (Fig. 16.3). In biliary atresia cases, it is expected that uptake of the radiotracer will be normal, and in approximately 80% of cases, there is no excretion of radiotracer into the intestine by 24 hours (Fig. 16.4).13 In contrast, with neonatal hepatitis, there will be very poor radiotracer extraction by the liver, but delayed images will show activity in the gastrointestinal tract (Fig. 16.5). For the diagnosis of biliary atresia, scintigraphy has a high sensitivity of 98% to 99% and a specificity of 68% to 72%.14,15 Differential considerations in the setting of nonvisualization of biliary structures include not only biliary atresia and neonatal hepatitis, but also congenital cholestatic syndromes such as progressive familial intrahepatic cholestasis, Alagille syndrome, and a cholangitic form of congenital hepatic fibrosis.16 Percutaneous liver biopsy and cholangiography may be performed for definitive diagnosis.
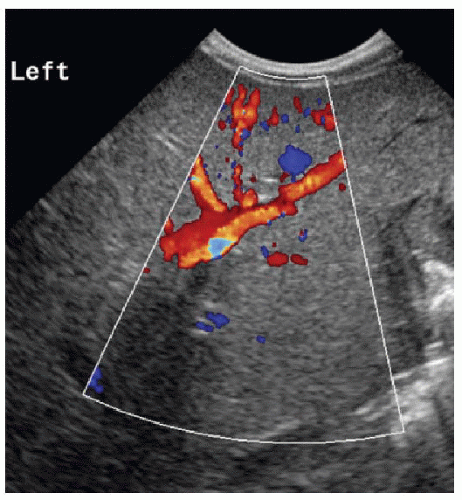 FIG. 16.2 • Biliary atresia. Color Doppler findings of subcapsular vessels in a 6-week-old male infant with biopsy-proven biliary atresia. |
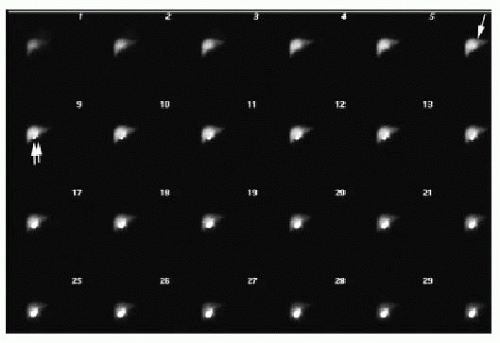 FIG. 16.3 • Normal scintigraphic hepatobiliary study. Technetium 99m mebrofenin injection through peripheral IV is followed by abdominal planar imaging with a frame rate of 60 seconds per frame. Prompt uptake of radiotracer by the liver with concomitant clearance from the blood pool (note lack of cardiac visualization) is normal. Intrahepatic bile ducts (arrow) are visualized by 5 minutes, and gallbladder activity is seen early (double arrows) by 8 minutes with gradual accumulation. Finally, enteric clearance of radiotracer is apparent from 35 minutes onward. |
 FIG. 16.4 • Biliary atresia on scintigraphic hepatobiliary study. Delayed 24-hour anterior, posterior, and lateral planar abdominal imaging is acquired after technetium-99m mebrofenin injection. Imaging performed on first day (not shown here) demonstrated prompt uptake of radiotracer by liver, followed by nonvisualization of intrahepatic ducts or gallbladder. Delayed imaging 1 day later fails to show accumulation of radiotracer in gallbladder. Vague excreted radiotracer is present in bilateral kidneys and bladder. Findings are consistent with biliary atresia or congenital cholestasis syndrome. Biopsy was pursued after imaging in this case, confirming biliary atresia. |
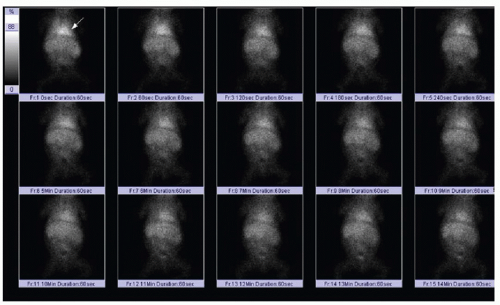 FIG. 16.5 • Idiopathic neonatal hepatitis on scintigraphic hepatobiliary study. Technetium 99m mebrofenin injection through peripheral IV is followed by abdominal planar imaging with a frame rate of 60 seconds per frame. In contrast to cases in Figures 16.3 and 16.4, there is incomplete clearance of radiotracer from the blood pool. Note persistent radiotracer within the cardiac blood pool (arrow), even beyond 10 minutes. |
CHOLEDOCHAL CYST
Choledochal cysts are rare congenital malformations of the biliary tree characterized by abnormal dilatation of all or part of the intra- and extrahepatic bile ducts. The incidence ranges from 1 in 13,000 to 1 in 2 million live births.17 Usually, these anomalies will be diagnosed in childhood, but approximately 20% are diagnosed in adults. They may be incidentally discovered on diagnostic imaging. The classic presenting symptoms of right upper quadrant pain, elevated bilirubin levels, and palpable mass are reported in up to 38% of pediatric cases.18 The pathogenesis of these cystic malformations is theorized to be secondary either to distal duct obstruction or to the effects of a long common pancreaticobiliary junction. The latter is an anatomic variant in which the main pancreatic duct and the common bile duct fuse outside the duodenal wall, with a common channel greater than 15 mm in length, allowing for pancreatic secretions to reflux into the biliary tree.19
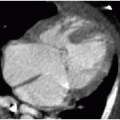
The pattern of bile duct dilatation is classified by Todani et al.20 (Fig. 16.6). Todani type I cysts involve only the extrahepatic duct, and these cysts may involve the entire extrahepatic duct (type Ia), may be focal (type Ib), or may have fusiform involvement of only the common bile duct (type Ic). Todani type II cysts are diverticula arising off of the extrahepatic duct. Todani type III cysts also only involve the common bile duct (like type Ic cysts), but these cysts are actually choledochoceles, reflecting ectasia of an intramural segment of the lower common bile duct within the duodenum. Todani type IV cysts are, by definition, multiple and may involve either just the extrahepatic bile duct (type IVb) or both the extrahepatic and intrahepatic bile ducts (type IVa). Depending on the location of the choledochal cysts, complications including cholelithiasis, hepaticolithiasis (in type IVa), pancreatitis, and portal hypertension may develop. There is also an increased incidence of malignancies including cholangiocarcinoma and gallbladder carcinoma in these patients.17,18 Diagnostic imaging modalities that may successfully demonstrate these anomalies include ultrasound, magnetic resonance and endoscopic cholangiopancreatography (MRCP and ERCP), and abdominal computed tomography. Given its high sensitivity for detecting ductal abnormalities, without requiring an invasive procedure, it is standard to attempt MRCP in cooperative patients before ERCP once suspicion of a choledochal cyst is raised on ultrasound (Fig. 16.7).21
Todani type V choledochal cysts are also multiple, but only seen in the large intrahepatic ducts. This disease, also known as
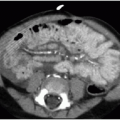
Caroli disease, is an autosomal recessive heritable disease that arises due to disordered development of the ductal plate in the biliary system, resulting in destructive inflammation and segmentally obstructed ducts.22,23 If small intrahepatic bile ducts are involved as well, the result is periductal hepatic fibrosis, and the combination is termed Caroli syndrome.23 Cross-sectional imaging will show scattered areas of intrahepatic segmental biliary duct dilatation (Fig. 16.8). Contrast-enhanced CT or MR imaging may show the “central dot” sign, which refers to punctate enhancement seen in the center of a dilated duct, reflecting portal venous branches in the areas of biliary tree involvement.24 Alternative diagnoses that might present with multiple areas of intrahepatic ductal ectasia or cysts include autosomal dominant polycystic liver disease, biliary hamartomas, biliary papillomatosis, and microabscesses.18,21 Caroli disease is associated with cholangiocarcinoma, renal cystic disease, and other choledochal cysts.23,25
Caroli disease, is an autosomal recessive heritable disease that arises due to disordered development of the ductal plate in the biliary system, resulting in destructive inflammation and segmentally obstructed ducts.22,23 If small intrahepatic bile ducts are involved as well, the result is periductal hepatic fibrosis, and the combination is termed Caroli syndrome.23 Cross-sectional imaging will show scattered areas of intrahepatic segmental biliary duct dilatation (Fig. 16.8). Contrast-enhanced CT or MR imaging may show the “central dot” sign, which refers to punctate enhancement seen in the center of a dilated duct, reflecting portal venous branches in the areas of biliary tree involvement.24 Alternative diagnoses that might present with multiple areas of intrahepatic ductal ectasia or cysts include autosomal dominant polycystic liver disease, biliary hamartomas, biliary papillomatosis, and microabscesses.18,21 Caroli disease is associated with cholangiocarcinoma, renal cystic disease, and other choledochal cysts.23,25
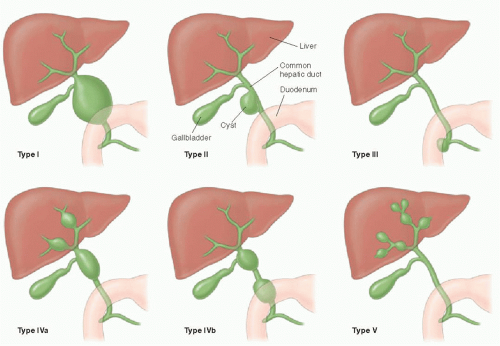 FIG. 16.6 • Schematic illustration of Todani classification system for choledochal cysts. |
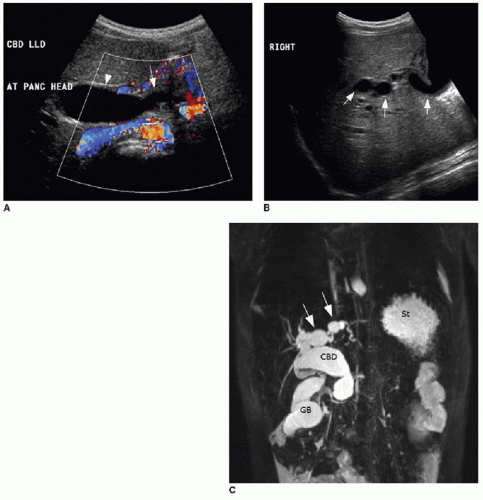 FIG. 16.7 • Type IVa choledochal cyst in a 4-year-old female with pancreatitis. Ultrasound images of the extrahepatic and intrahepatic bile ducts show abnormal dilatation. The lower aspect of the common bile duct (arrow, image A) rapidly dilates abnormally (arrowhead). B: Variable intrahepatic saccular ductal dilatation (arrows) is observed. C: Magnetic resonance cholangiopancreatography (MRCP) using heavily T2-weighted maximum projection images shows the extent of abnormal ductal dilatation. Arrows mark left intrahepatic ductal dilatation. (CBD, common bile duct; GB, gallbladder.) |
Choledochal cysts are generally treated by surgical excision. Mucosa-to-mucosa biliary flow is achieved following excision with biliary-enteric anastamosis, such as by hepaticojejunostomy with a Roux-en-Y after extrahepatic cyst resection.26 A type III choledochal cyst can be treated endoscopically with sphincterotomy and unroofing of the choledochocele.27 Extensive intrahepatic choledochal cysts cannot be completely excised; however, if focal or regional, partial hepatectomy is the best surgical option.26 Complete excision of the extrahepatic duct with a wide hilar hepaticojejunostomy has been reported to reduce the likelihood of late complications such as cholangitis, pancreatitis, and stone formation.28 End-stage liver disease and portal hypertension develop secondary to hepatic fibrosis, and liver transplantation may be appropriate in these high-risk patients.29
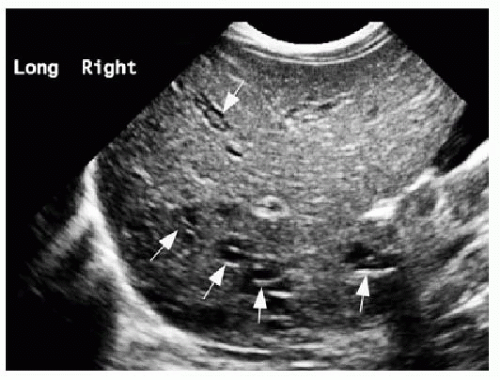 FIG. 16.8 • Caroli syndrome. Seven-month-old infant with autosomal recessive polycystic kidney disease (ARPCKD) undergoes ultrasound evaluation. Central dot sign is observed, indicating intrahepatic ductal dilatation secondary to congenital malformation of the ductal plate (associated with ARPCKD). Arrows mark multiple markedly dilated ducts, each containing internal echogenic dot, reflecting the small portal venous branches surrounded by ductal dilatation. The CT equivalent of the central dot sign shows a central dot of enhancement within dilated ducts, representing portal radicles. |
HEPATIC TUMORS
Malignant and benign liver tumors are relatively uncommon in the pediatric population compared with tumors arising in other systems. However, these masses arise in all pediatric age groups, from the fetus through the young adult, and they may present diagnostic challenges for any practicing radiologist.30, 31 and 32 As with adults, the most common type of malignancy found in the liver of children is metastatic disease—most commonly neuroblastoma, Wilms tumor, and lymphoma. Of the primary hepatic tumors found in children, approximately two-thirds of cases are malignant, and these constitute 1% to 2% of all pediatric cancers.31 An extensive review of over 700 pediatric tumors30 summarized the most common types of intrahepatic pediatric malignant tumors, which are as follows, in decreasing order of frequency: hepatoblastoma, hepatocellular carcinoma, undifferentiated embryonal sarcoma, angiosarcoma, and embryonal rhabdomyosarcoma. The most common benign tumors include congenital hemangioma, nodular regenerative hyperplasia, mesenchymal hamartoma, focal regenerative nodule, and hepatic adenoma.
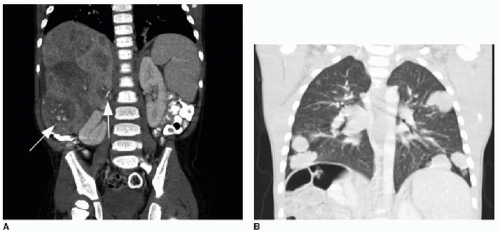 FIG. 16.9 • Hepatoblastoma. Two-year-old boy was evaluated for abdominal mass with contrast-enhanced abdominal and pelvic CT study. A: Coronal reconstruction shows a large intrahepatic mass with multilobulated contours, heterogeneous enhancement, and intratumoral punctate calcifications (arrows). B: One year following surgical resection and chemotherapy, disease recurrence manifested with pulmonary metastases. |
Hepatoblastoma, the most common pediatric hepatic malignancy, is diagnosed primarily in young children, with approximately 70% diagnosed by age 2 years and 90% by age 5 years.30,33




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
