1. Recognize typical imaging features of Wilms tumor, including common sites for metastases.
2. Describe the surgical staging of Wilms tumor.
3. Identify nephroblastomatosis on imaging, and discuss its implications.
4. List at least one distinguishing imaging or clinical feature (e.g., patient demographic) for each of the following renal tumors: clear cell sarcoma, multilocular cystic tumor, mesoblastic nephroma, renal cell carcinoma, renal lymphoma, and angiomyolipoma.
5. Recognize typical imaging features of neuroblastoma, including three common sites for the primary tumor.
6. List at least three ways to differentiate neuroblastoma from Wilms tumor by imaging.
7. Identify adrenal hemorrhage on ultrasound.
8. Describe at least one typical imaging or clinical feature of the following pelvic tumors: rhabdomyosarcoma and sacrococcygeal teratoma.
INTRODUCTION
The tumors presented in this chapter account for a large proportion of palpable abdominal masses in young children. Wilms tumor and neuroblastoma are among the most common childhood neoplasms overall. Nevertheless, a variety of lesions may arise within the kidney, adrenals, and pelvis, and one must remain open-minded when evaluating these regions. Nonspecific symptoms such as abdominal pain and distension may prompt initial imaging with radiographs. US, CT, or MR are needed for further tumor characterization and staging. Close collaboration among the radiologist, oncologist, and surgeon is always warranted during imaging interpretation and therapy planning.
RENAL TUMORS
Wilms tumor
Wilms tumor is among the most common abdominal malignancies of childhood and constitutes nearly 90% of all pediatric renal tumors.1,2 Its peak incidence is at 3 to 4 years of age, and 80% of patients present before 5 years of age.1,2,3and4 The majority of children present with a palpable mass, abdominal distension, or pain. Wilms tumors arise from nephrogenic rests, which are foci of embryonic precursor kidney cells that persist postnatally (see next section “Nephroblastomatosis”). Postmortem studies have found nephrogenic rests in 1% of infants who died of other causes and in 30% to 40% of infants with Wilms tumors.5,6 The vast majority of Wilms tumors arise from a single kidney; bilateral tumors are present in 4% to 13% of children.2,3 Bilateral disease occurs almost exclusively in cases of nephroblastomatosis.
Wilms tumor has been linked to two loci on chromosome 11. WT1 gene (11p13) mutations are associated with WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation) and Denys-Drash syndrome. WT2 gene (11p15) mutations are responsible for Beckwith-Wiedemann syndrome and hemihypertrophy.1,2,5,7,8 Familial cases account for less than 5% of children with Wilms tumor. They have an earlier age of onset and increased frequency of bilateral lesions.7,8
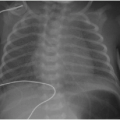
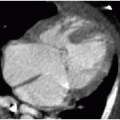
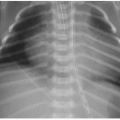
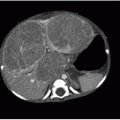
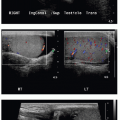
On US, Wilms tumors exhibit heterogeneous echogenicity and variable perfusion on color Doppler imaging (Fig. 23.1). They have a propensity for venous invasion, so evaluation for tumor thrombus within the renal vein and IVC is warranted (Fig. 23.2). Within the tumor, there may be anechoic or hypoechoic cystic necrosis, or hemorrhage. On contrastenhanced CT, Wilms tumor is typically a spherical hypodense mass that distorts the surrounding renal parenchyma and collecting system (Figs. 23.1, 23.2 and 23.3). Fat, hemorrhage, necrosis, and calcification may be identified. Wilms tumor often grows like a solid sphere or ball, displacing adjacent structures such as vessels and bowel. This propensity to displace rather than encase vessels is an important distinguishing feature of Wilms tumor, in contradistinction to neuroblastoma (refer to “Neuroblastoma” section). Dystrophic calcification is seen in about 10% of lesions—far less frequent than in neuroblastoma. The “claw sign” with Wilms tumor refers to strips of parenchyma that extend around the mass, confirming its renal origin (Fig. 23.1B). On MR, Wilms tumors are generally T1W iso- to hypointense, T2W hyperintense, and enhance less than the adjacent renal parenchyma. Treated tumors may show hypointense T2W signal. The masses are often well defined with a pseudocapsule, but have the potential to transgress the renal capsule and directly invade the mesentery or seed the peritoneal cavity. The lungs are the most common site of metastatic disease. Ten to twenty percent of children have lung metastases at time of presentation. Additional metastatic locations include the liver and regional lymph nodes. Skeletal metastases are atypical for Wilms tumors, and bone scans are not generally performed. The vast majority of lesions are FDG avid on PET-CT.1,2and3,7,9,10and11
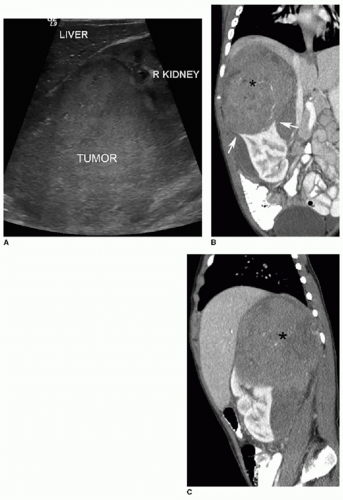
FIG. 23.1 • Wilms tumor in a 2-year-old male. A: Sagittal US image demonstrates a heterogeneously hyperechoic mass arising from the superior portion of the right kidney. B,C: Coronal (B) and sagittal (C) images from a contrastenhanced CT shows a large hypodense mass (* on both B and C) engulfing and distorting the right kidney. The “claw sign” (arrows in B) refers to parenchymal strips outlining a portion of the tumor, confirming its renal origin.
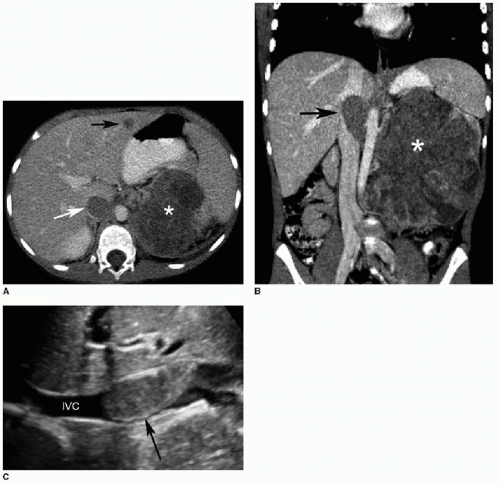
FIG. 23.2 • Wilms tumor with venous invasion in a 7-year-old female. A: Axial contrast-enhanced CT image reveals a large heterogeneously hypodense mass within the left kidney (*). Note the tumor thrombus within the IVC (white arrow) and the metastasis in the left hepatic lobe (black arrow). B: Coronal image shows the primary renal tumor (*) and the tumor thrombus in the IVC (arrow). C: Longitudinal US image illustrates the IVC thrombus (arrow). Color Doppler (not shown) demonstrated internal vascularity within this lesion, confirming tumoral extension rather than a bland thrombus.
In the United States, staging for Wilms tumor is determined surgically (Table 23.1). In stage 1 disease, the tumor is limited to the kidney and has been completely resected. In stage 2, the tumor extends beyond the renal capsule but is completely resected. In stage 3 disease, there is residual tumor after surgery that is confined to the abdomen. Stage 4 disease involves either hematogenous metastases or lymph nodal metastases outside of the abdomen or pelvis. Stage 5 is bilateral Wilms tumors (Fig. 23.3).12,13
The standard treatment for unilateral Wilms tumor is radical nephrectomy. En bloc resection of the tumor is highly desirable, as tumor spillage results in a sixfold increase in local abdominal recurrence.7 Preoperative chemotherapy is used for unresectable bulky masses, for bilateral Wilms tumors, and when tumor thrombus extends into the right atrium from the IVC.10 The prognosis for Wilms tumor is generally favorable. Long-term survival exceeds 90% for children with localized disease and is greater than 70% for patients with metastases.1
Nephrogenic rests/nephroblastomatosis
The kidneys begin developing by 5 weeks of fetal life and are fully developed by the 36th week of gestation. Nephrogenic rests are foci of embryonic renal parenchyma (metanephric blastema) that persist beyond 36 weeks gestational age. Multiple or diffuse rests are referred to as nephroblastomatosis.2,3,5 Nephrogenic rests are found in 1% of healthy neonates and are usually absent by 4 months of age. They are asymptomatic and are discovered either incidentally when imaging the abdomen for another purpose or in conjunction with a Wilms tumor.3 The potential for these renal precursor cells to undergo malignant degeneration in Wilms tumor is why they garner attention from oncologists and radiologists. Nephrogenic rests may be categorized by their location within the kidney as either perilobar or intralobar. While perilobar rests are much more common, intralobar rests are more likely to degenerate into Wilms tumor.1 Nearly all cases of bilateral Wilms tumors arise from preexisting nephroblastomatosis.
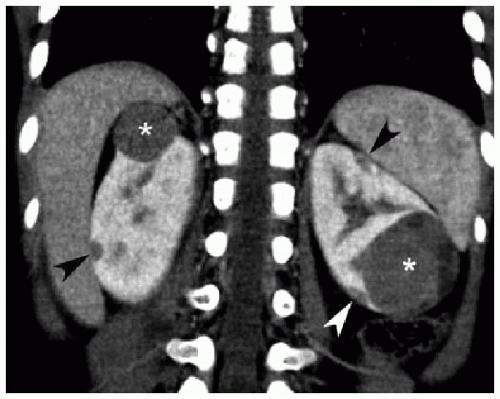
FIG. 23.3 • Bilateral Wilms tumor in a 3-year-old female. Coronal contrast-enhanced CT image exhibits spherical hypoenhancing parenchymal masses (*) in both kidneys. There are also scattered subcapsular ovoid hypodensities (arrowheads) representing nephrogenic rests. Nearly all patients with bilateral Wilms tumors have underlying nephrogenic rests/nephroblastomatosis.
On ultrasound, nephrogenic rests may be either hypoechoic or isoechoic to the surrounding normal parenchyma (Figs. 23.4 and 23.5). In the perilobar form, multiple peripheral, hypoechoic masses may be seen. Contrast-enhanced CT is more sensitive and specific for this condition. Diffuse perilobar nephroblastomatosis manifests as confluent hypodense masses along the periphery of the kidney (Figs. 23.4 and 23.5). The rests are distinguished from the more avidly enhancing surrounding parenchyma, and the affected kidney is often enlarged and distorted. On MR, compared to normal renal parenchyma, nephrogenic rests are T1Wisointense, T2W slightly hyperintense and show less enhancement. On all modalities, concern for Wilms tumor transformation should be raised when one or more of these masses enlarges rapidly or becomes more heterogeneous in appearance.1,2and3,10,14
Most cases of nephroblastomatosis spontaneously regress. Currently, there is no consensus guideline regarding imaging surveillance of nephroblastomatosis. It is common practice to perform a CT at time of diagnosis, usually when the child is less than 1 year old, and renal US every 3 months until 8 years of age. If there is suspicion for malignant degeneration on ultrasound, then contrast-enhanced CT or MR is performed.1,7,10,15
Clear cell sarcoma of the kidney
Clear cell sarcomas constitute 5% of pediatric renal tumors. It used to be considered a subtype of Wilms tumor, but was subsequently reclassified as a distinct lesion because of its unique histologic and biologic features. Clear cell sarcomas present as palpable abdominal masses in children aged 1 to 4 years, and there is a 2:1 male predilection. Virtually all cases are unilateral. On imaging, clear cell sarcomas appear as heterogeneous solid renal masses that are very similar to Wilms tumors (Fig. 23.6). In contrast to Wilms, clear cell tumors have a tendency to metastasize to the skeletal system. Therefore, bone scintigraphy or radiographic skeletal surveys are warranted. Treatment consists of radical nephrectomy and chemotherapy. Clear cell sarcoma is more aggressive than Wilms tumor and carries increased rates of recurrence and mortality. Long-term survival rates are 60% to 70%.1,2,7,16,17and18
Table 23.1 CHILDREN’S ONCOLOGY GROUP STAGING OF WILMS TUMOR
Stage 1
•
Completely resected tumor limited to the kidney with intact capsule
•
No biopsy or rupture of tumor prior to removal
•
No involvement of vessels or renal sinuses
•
No tumor at or beyond margins of resection
•
Regional lymph nodes negative for tumor
Stage 2
•
Completely resected tumor
•
No tumor at or beyond margins of resection
•
Regional lymph nodes negative for tumor
•
One or more of the following findings:
• Penetration of the renal capsule
• Invasion of vasculature extending beyond renal parenchyma
Stage 3
•
Residual tumor present after surgery, confined to the abdomen, with one or more of the following findings:
• One or more regional lymph nodes positive for tumor
• Tumor implanted on, or penetrating through, the peritoneum
• Presence of gross unresected tumor or tumor at margin of resection
• Any tumor spillage occurring before or during surgery, including biopsy
• Tumor removed in more than one piece
Stage 4
•
Presence of hematogenous metastases (e.g., lung, liver, bone, brain)
•
Presence of lymph node metastases outside the abdomen or pelvis
Stage 5
•
Wilms tumor in both kidneys
From Bates DG, Feinstein KA. Renal neoplasms. In: Coley BD, ed. Caffey’s Pediatric Diagnostic Imaging. 12th ed. Philadelphia, PA: Elsevier Saunders; 2013:1218-1226; Gratias EJ, Dome JS. Current and emerging chemotherapy treatment strategies for Wilms tumor in North America. Paediatr Drugs. 2008;10(2):115-124.
Multilocular cystic renal tumor
These lesions include multilocular cystic nephroma, a benign lesion containing mature fibrous septa and tubules, and cystic partially differentiated nephroblastoma, which contains foci of renal blastema cells within the septa. Multilocular cystic tumors are distinguished from cystic Wilms tumors pathologically by the lack of solid nephroblastomatous tissue. They have an unusual bimodal demographic presentation—young boys aged 3 months to 4 years, and middle-aged women. Multilocular cystic renal tumors are nonfamilial, are not associated with cystic disease in other organs, and are only sporadically linked with other congenital anomalies. They present as a palpable abdominal mass in a male infant or toddler.1,2,10,19
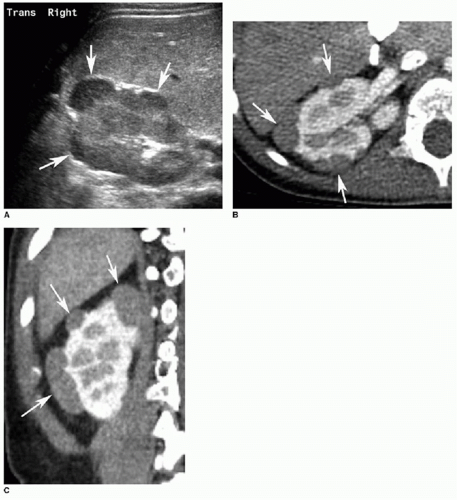
FIG. 23.4 • Nephrogenic rests in a 2-year-old male. A: Transverse US image of the right kidney illustrates multiple peripheral, hypoechoic ovoid masses (arrows). B,C: Axial (B) and coronal (C) contrast-enhanced CT images reveal peripheral hypoenhancing masses (arrows in both B and C).
On all imaging modalities, multilocular cystic renal tumors appear as well-defined masses containing cysts of variable size (Fig. 23.7). The cysts are typically anechoic on US, fluid density on CT, and T2W-hyperintense on MR. When the cysts are small and numerous, the lesion may take on a solid appearance on US because of the acoustic interfaces, similar to autosomal recessive polycystic kidney disease. Hemorrhage and calcification may be seen with these tumors. Septations are also of variable thickness. Postcontrast imaging generally shows only peripheral and septal enhancement (Fig. 23.7B). Large masses will show the “claw sign,” confirming renal origin and may herniate into the renal pelvis.1,2,7,10,19-21
While multilocular cystic renal tumors are benign, differentiating these histopathologies from more aggressive ones such as cystic Wilms tumor is not possible by imaging. Therefore, these patients undergo complete or partial nephrectomy. Surgery is typically curative, and the prognosis is very favorable.19,20and21
Only gold members can continue reading. Log In or Register to continue