Definition A facial cleft involving the upper lip or palate, usually occurring to the left or right of midline, the cleft lip or cleft palate may occur as an isolated malformation or as part of a multiple-malformation syndrome. Midline facial clefts may be associated with underlying brain malformations, especially holoprosencephaly.
Epidemiology Occurrence is 1 in 1000 births (M2:F1) for cleft lip or cleft palate and 0.65 in 1000 births for isolated cleft palate (M2:F3). There is marked ethnic and racial variation in incidence (Caucasian 1 to African American 0.6). Some studies suggested that maternal preconceptional folic acid supplementation decreases the incidence of nonsyndromic clefts. Medial facial clefts account for less than 1% of all facial clefts.
Embryology The primary palate (anterior to incisive foramina) and secondary palate (posterior to the incisive foramina) are embryologically distinct. The upper lip and primary palate have usually fused by the seventh week of gestation. Formation of the secondary palate occurs by fusion of the palatal shelf by the 12th week. Cleft lip and cleft palate are due to a failure of union of the frontonasal process of the face with the lateral maxillary prominences at about 7 weeks’ gestation. Approximately 60% of cases are isolated. Almost 300 multiple-malformation syndromes have been described with cleft lip or cleft palate. Chromosomal abnormalities may be found in 3.9% of isolated cleft lip and palate and in up to 63% when associated anomalies are present. A variety of chromosomal abnormalities have been implicated, including the 22q11 deletion. The most common associated anomalies are those involving the lumbar and cervical spine (33%) and heart (24%).
Midline facial clefts are the result of a deficient frontonasal development process that is normally induced by the underlying brain. Midline facial clefts with underlying brain abnormalities are seen in trisomy 13, as well as other chromosomal abnormalities and genetic syndromes.
Inheritance Patterns Most isolated cleft lips or cleft palates show multifactorial inheritance, but up to 20% are a component of dominant, recessive, and X-linked syndromes (including van der Woude syndrome). Determining the pattern of inheritance depends on an accurate diagnosis.
Teratogens Alcohol, smoking, maternal phenylketonuria, hyperthermia, hydantoin, trimethadione, aminopterin, carbamazepine, valproate, retinoic acid, and methotrexate are teratogenic.
Prognosis The prognosis for a good cosmetic and functional repair with isolated cleft lip or cleft palate is excellent. Otherwise, the prognosis is dependent on any associated malformations or a syndrome diagnosis. Midline clefts, if they are associated with underlying brain malformations, usually carry a poor prognosis.
Unilateral cleft lip with or without cleft palate
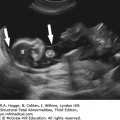
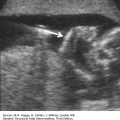
A space is identified in the soft tissue of the lip and may be seen just on the inferior surface of the lip (incomplete cleft) versus into the nostril (complete cleft).
The alae of the nose are often inferiorly displaced with a complete cleft.
The hard palate (alveolar ridge) can often be imaged just posterior to the lip, and a defect in this region appears as a space between the tooth buds.
Bilateral cleft lip with or without palatal defect
Bilateral cleft lip is almost universally found in the setting of a bilateral cleft palate as well.
There are soft tissue clefts seen on each half of the upper lip.
The bilateral clefting in the palate causes the central alveolar ridge segment to lose its bony attachment to the remainder of the maxilla, resulting in an anterior displacement of the lip and palate. This can be seen sonographically as a premaxillary protuberance that is characteristic of bilateral cleft lip and palate.
Isolated cleft palate or cleft soft (secondary) palate
The presence of isolated cleft palate (not associated with cleft lip) or cleft soft (secondary) palate is difficult to identify prenatally.
This diagnosis is most commonly established in the neonatal period.
Median cleft lip and palate
There is an absence of the central maxilla and upper lip with a deformed nose that may be absent or replaced by a proboscis.
The facial profile is often flat.
This type of cleft lip and palate is almost always associated with other facial abnormalities, such as hypotelorism or cyclopia, and brain abnormalities, particularly holoprosencephaly.
Atypical facial clefts
Typically, these are the result of amniotic bands.
The appearance of the lip and face is quite variable, although often defects seen in this setting are severe.
Close evaluation for additional fetal deformations related to amniotic bands is warranted.
Three-dimensional (3-D) sonography has been demonstrated to more accurately diagnose facial clefting anomalies. The 3-D surface view can more clearly demonstrate a more life-like appearance of the defect, which is key in confirming the diagnosis and counseling the family.
The presence of cleft lip and palate can be identified as early as the late first trimester, particularly when transvaginal imaging is performed.
Bilateral cleft lip and palate may be mistaken for a facial mass, such as a teratoma or proboscis.
It may be difficult to accurately image the lip and palate in a fetus that is in a prone position facing the maternal back or when image quality is poor secondary to large maternal body habitus.
With 3-D sonography, movement artifacts, or overlapping fetal parts, may mimic a facial cleft.
Epignathus (facial teratoma)
The mass is asymmetrical and enters the mouth.
There is no associated soft tissue cleft.
Investigations and Consultations Required Chromosome studies, with microarray, should be done in all cases, including apparently isolated cleft lip/palate. Because of the strong association with cardiac abnormalities, fetal echocardiography is an important component of the evaluation. The parents should be examined by a pediatric dysmorphologist for possible genetic disorders that are inherited in an autosomal dominant fashion. Coordinated prenatal care with a fetal anomaly center, genetics, neonatology, and pediatric plastic surgery is recommended.
Monitoring Because of the significant risk of other abnormalities that may not be detected on the initial sonographic evaluation, serial ultrasound is recommended. A follow-up ultrasound examination at the beginning of the third trimester, with 3-D assessment, may provide a more accurate surface image of the fetal face, which can be helpful for counseling.
Pregnancy Course No specific obstetric complications should be expected in the fetus with cleft lip/palate. Mild polyhydramnios may occur occasionally.
Pregnancy Termination Issues An intact fetus is necessary for pathologic assessment to confirm the ultrasound findings and to assess for additional anomalies that can provide a precise syndrome diagnosis.
Delivery Delivery should occur at a center experienced in taking care of neonates with facial clefting. A medical geneticist should also be available to assess for additional dysmorphic features or genetic syndromes.
Resuscitation Fetal distress is not expected with isolated facial clefting. However, infants found to have additional anomalies often need additional support, including respiratory assistance. The decision to intervene is based on the prognosis for the specific syndrome or anomaly complex.
Transport Referral to a tertiary center following birth is not indicated for isolated facial clefts unless a satisfactory feeding technique cannot be established. With multiple anomalies, referral for more extensive diagnostic evaluation is appropriate.
Testing and Confirmation Assessment of the type, and extent, of the cleft is recommended following delivery. Because clefts in the soft palate cannot typically be identified antenatally, a careful assessment of the lip and hard and soft palate is recommended. Because of the increased prevalence of associated anomalies, which may not have been detected antenatally, a thorough neonatal evaluation for additional anomalies is warranted.
Nursery Management Establishing a successful oral feeding technique and facilitating parental adaptation are the initial objectives in management. There are multiple special devices available for use, and use of a customized prosthetic device for large palatal defects may help some infants. Referral to a center with a multidisciplinary orofacial team for long-term management, including surgical repair and rehabilitation, is essential.
Surgical Reconstructive Procedures All patients with orofacial clefts will require surgical interventions, which may continue until they reach skeletal maturity. Surgical procedures, and timing, are based on the child’s diagnosis and the presence of any comorbid conditions.
Primary Lip/Nose Reconstruction Incomplete cleft lips and less-severe complete cleft lips are repaired generally between 3 and 6 months of age. The primary repair includes approximation of the lip elements, including muscle and skin, as well as the nasal cartilages and tissues. For more severe, or wide complete, clefts of the lip and alveolus, treatment may involve staged interventions.
Primary Palate Repair Children with cleft palate generally undergo primary palatoplasty, or palate repair, between 9 and 18 months of age. Palatoplasty is necessary for a child to develop normal speech and velopharyngeal competence.
Maxillary Alveolar Cleft Reconstruction Children who are born with complete cleft lip with alveolar involvement will undergo reconstruction of their alveolar defect between 6 and 10 years of age.
Surgical Results/Prognosis Surgery usually restores facial aesthetics and functional speech. The rate of complications from cleft palate closure, requiring late palatal lengthening or fistula closure, ranges from 8% to 20%. The more extensive the deformities are, the greater the number of procedures that will be required to achieve the desired results. Surgical procedures may extend into the end of the craniofacial growth period in the teenaged years.
FIGURE 7.1A-D
Sonographic images of normal face: (A) axial view of intact hard palate with tooth buds (arrow); (B) coronal view demonstrating tip of nose (triangle), nostrils, upper lip (arrow), tongue, and lower lip (arrow); (C) sagittal view demonstrating normal fetal profile; (D) 3-D surface rendering of superficial facial features.













Definition Nuchal cystic hygromas (CHs) are characterized by single or multiple congenital cysts of the lymphatic system, most commonly found within the soft tissues of the posterior and lateral neck.
Epidemiology Incidence is 1 in 285 first-trimester fetuses.
Embryology Nuchal cystic hygromas are the clinical consequence of a delay in development, or absence, of the communications that normally develop between the jugular lymph sacs and the internal jugular veins at approximately 5-6 weeks’ gestation. Failure of communication between the lymphatic and venous systems leads to enlarged lymphatic sacs and channels. They are most commonly seen in the subcutaneous region of the posterior fetal neck but may also extend to the lateral neck. As the pressure within the lymphatic system increases, lymphatic fluid can be visualized within the subcutaneous tissue, the pleural space, and the pericardial space and within the abdominal cavity (nonimmune fetal hydrops), which is frequently lethal. Late manifestations of the cystic hygroma, if the fetus survives, include redundancy of posterior nuchal skin, neck webbing, and elevation and anterior rotation of the ears. Occasionally, the cystic hygroma is only unilateral. These cases are generally not associated with chromosomal abnormalities or other genetic conditions.
Inheritance Patterns Multiple-malformation syndromes with cystic hygromas include multiple-pterygium syndrome (autosomal recessive, X linked); Noonan syndrome (autosomal dominant); Roberts syndrome (autosomal recessive); as well as others. Chromosomal abnormalities associated with cystic hygroma include Turner syndrome; trisomies 13, 18, and 21; Klinefelter syndrome, and other aneuploidies.
Teratogens No teratogens are reported.
Prognosis Almost all fetuses with cystic hygroma and hydrops die antenatally. Survivors with lymphatic recanalization may present with a webbed neck or redundant nuchal skin. Even for chromosomally normal fetuses that survive, the prevalence of congenital anomalies, some of which may not be identifiable antenatally, is significantly increased.
Cystic hygromas present as septated fluid collections in the subcutaneous tissue of the posterior fetal neck.
They may extend laterally to the sides of the neck and inferiorly down the fetal back.
In the second trimester, they are often large and can exceed the size of the fetal calvarium.
Skin thickening
Secondary to subcutaneous lymphedema
Often develops in the second trimester but can also be seen in the late first trimester
Presents as diffuse anasarca, which can be present throughout the fetal skin
Hydrops
With large hygromas, fetal nonimmune hydrops often develops.
The presence of nonimmune hydrops in a fetus with a cystic hygroma is a particularly ominous sign, and fetal death often occurs within several weeks.
Oligohydramnios in cases of hydrops is less frequent.
Often, cystic hygromas are first identifiable in the middle of the first trimester (after 10 weeks’ gestation).
Because small cystic hygromas can spontaneously disappear, patients who had cystic hygromas early on may later appear normal and yet have syndromes, such as Turner or Down syndrome, at birth.
The resolution of a cystic hygroma does not eliminate the risk for genetic syndromes or aneuploidy.
Thickened nuchal translucency (NT) may have a similar appearance.
A simple NT is confined to the occipital region and does not contain septations.
As the size of the fluid collection behind the neck increases, the distinction between a thickened NT and a cystic hygroma is lost.
The association with an abnormal karyotype, or other structural abnormalities, is the same for an NT above the 99th centile (3.5 mm) and a cystic hygroma.
Encephalocele or meningocele
The mass is seen focally at the posterior occiput or midline upper spine.
Both cases almost always also demonstrate abnormal intracranial findings.
Unfused amniotic membrane
Before 13 weeks, an unfused amniotic membrane may lie adjacent to the fetus and be confused with a cystic hygroma.
Lymphangioma
Lymphangioma may have a similar multiseptated appearance.
However, a lymphangioma is typically unilateral and develops later in pregnancy.
Investigations and Consultations Required Approximately 50% of fetuses with a cystic hygroma will be found to have a chromosomal abnormality. The most common chromosomal abnormalities identified are trisomy 21, 45X, and trisomy 18. Noonan syndrome may account for up to 15% of cases. A gene panel for genes associated with Noonan syndrome should be done if chromosome analysis and microarray studies are negative. For those fetuses found to be euploid, up to 30% will have additional anomalies, most commonly affecting the cardiac, skeletal, and genitourinary systems. A detailed anatomic survey in midgestation, as well as a fetal echocardiogram, is recommended. Only approximately 15% of fetuses found to have a cystic hygroma will be born alive, with no long-term sequelae.
Fetal Intervention In utero drainage procedures have no role in management. Spontaneous resolution may occur, although is difficult to predict. Progression to fetal hydrops suggests impending fetal death in most cases.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


