Neuroendocrine Hyperplasia and Pulmonary Tumorlets
Etiology, Prevalence, and Epidemiology
Normal lung tissue contains scattered neuroendocrine (Kulchitsky) cells within the bronchial and bronchiolar epithelium. These cells play a role in the detection of hypoxia as well as fetal lung development and may be involved in local epithelial cell growth and regeneration. Hyperplasia of these cells can be seen as a response to chronic airway inflammation, such as in patients with emphysema or chronic bronchitis. When no known cause can be identified, this hyperplasia is designated diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH). Extension of hyperplastic neuroendocrine cells beyond the basement membrane is termed a tumorlet if the cell mass is less than 5 mm and carcinoid tumor if the cell mass is 5 mm or greater in diameter. Neuroendocrine cell hyperplasia and tumorlets may coexist with carcinoid tumors. In the absence of lung injury (leading to reactive pulmonary neuroendocrine cell hyperplasia), DIPNECH is considered a preinvasive condition that may develop into carcinoid tumors, and imaging follow-up is required.
DIPNECH typically manifests in the fifth to sixth decades of life, with women affected nearly four times as often as men. DIPNECH usually occurs in nonsmokers, but former and current smokers are occasionally affected. Tumorlets also demonstrate a female preponderance (>4 : 1) and are often seen in patients 60 to 70 years old.
In the 2015 update to the World Health Organization (WHO) classification of lung tumors, DIPNECH is classified as a preinvasive lesion. Although the diagnosis remains purely histologic, some authors have proposed the term DIPNECH syndrome be used to indicate patients with respiratory symptoms and appropriate imaging findings. Although initially thought to be rare, with few cases described in the literature, DIPNECH is likely an underrecognized condition.
Clinical Presentation
Up to half of patients with DIPNECH or tumorlets are asymptomatic, and the diagnosis is made incidentally on chest computed tomography (CT) or surgical lung biopsy performed for other reasons. When patients are symptomatic, they often present with a history of protracted nonproductive cough and/or dyspnea and are often mistakenly diagnosed with asthma. Pulmonary function tests may show obstructive or obstructive/restrictive alterations, likely caused by airway obstruction from the hyperplasia as well as constrictive bronchiolitis from peribronchial fibrosis.
Pathophysiology
Neuroendocrine cell hyperplasia (NECH) is characterized by single, clustered, or linear arrays of Kulchitsky cells confined by the basement membrane. Tumorlets are distinguished by extension beyond the basement membrane. Both of these entities show no mitoses or necrosis per 10 high-power fields on histologic examination. NECH and tumorlets are usually an incidental finding in lung parenchyma scarred by bronchiectasis or other chronic inflammatory processes.
As stated earlier, constrictive bronchiolitis in patients is secondary to submucosal fibrosis of affected airways in regions with and regions without tumorlets and NECH.
Imaging Findings
Radiography
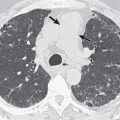
Chest radiographs of patients with DIPNECH or pulmonary tumorlets may demonstrate pulmonary micronodules ( Fig. 19.1 ) but are more often normal. Secondary signs of constrictive bronchiolitis, including hyperinflation and attenuation of peripheral pulmonary vasculature, can occasionally be seen.

Computed Tomography
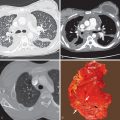
High-resolution CT of the chest with inspiratory and expiratory imaging will demonstrate multifocal pulmonary micronodules with or without associated mosaic attenuation or air trapping. These micronodules are typically in a centrilobular distribution and can be ground-glass or solid in attenuation. Nodules less than 5 mm correspond to carcinoid tumorlets, whereas nodules 5 mm or greater represent carcinoid tumors at histopathologic examination. Asymptomatic patients are more likely to demonstrate micronodules without significant mosaic attenuation as the extent of mosaic attenuation often correlates with physiologic airflow obstruction. Bronchial wall thickening with or without bronchiectasis is occasionally seen. Nodular bronchial wall thickening is the most direct radiologic-pathologic correlation of NECH, corresponding to the histologic findings of intraluminal protrusion of the proliferating cells. There are no imaging features to distinguish NECH from pulmonary tumorlets. This distinction can only currently be made on histology. A recent study, evaluating 30 patients with DIPNECH, proposed nonsurgical diagnostic criteria. These included clinical presentation, pulmonary function, imaging findings on high-resolution CT, findings from transbronchial biopsy, and serum markers such as elevated serum chromogranin A levels.
Differential Diagnosis
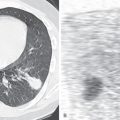
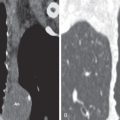
Detection of one or more pulmonary nodules is a common diagnostic dilemma on CT. The differential considerations for a single nodule or multiple pulmonary nodules include infectious, inflammatory, and neoplastic etiologies. The diagnosis of DIPNECH may be suggested based upon typical imaging findings, mosaic attenuation of the lung parenchyma on inspiratory imaging ( Fig. 19.2 ), and air trapping on expiratory imaging. Definitive diagnosis, however, requires open lung or thoracoscopic biopsy ( Fig. 19.3 ).


Synopsis of Treatment Options
Asymptomatic patients are usually managed conservatively, with a good long-term prognosis of 83% survival at 5 years. Long-term imaging follow-up is recommended to exclude nodule growth, development of carcinoid tumors, or metastatic disease. Patients presenting with worsening lung function can be managed more aggressively, with some showing a response to steroids, interferon-α, or chemotherapeutic agents. The use of somatostatin receptor analogues (given the neuroendocrine origin of cells) has shown good results with limited side effects.
- •
Neuroendocrine cell hyperplasia represents proliferation of Kulchitsky cells within the bronchial and bronchiolar epithelium.
- •
Pulmonary tumorlets are nodular proliferations of these same cells with extension beyond the basement membrane. By definition, they measure less than 5 mm in diameter; if 5 mm or greater, they are referred to as carcinoid tumors.
- •
Most patients are asymptomatic; however, when symptoms are present, they are nonspecific and are most commonly a history of protracted nonproductive cough and/or dyspnea that is often initially mistaken for asthma.
- •
Chest radiographs are predominantly normal but may show increased lung volumes.
- •
Inspiratory CT may show mosaic attenuation (usually in symptomatic patients), and expiratory CT may show air trapping.
- •
New WHO classification categorizes diffuse idiopathic pulmonary neuroendocrine cell hyperplasia as a preinvasive entity, and long-term imaging follow-up is recommended to exclude nodule growth, development of carcinoid tumors, or metastatic disease.
Typical and Atypical Carcinoid Tumors
Etiology, Prevalence, and Epidemiology
The respiratory tract is the second most common location for carcinoid tumors, accounting for 10% to 30% of carcinoids. Pulmonary carcinoids have shown an increase in prevalence relative to other primary sources over the past few decades. Carcinoid tumors constitute about 1% to 2% of all primary lung malignancies. As noted previously, carcinoid tumors represent an extension of the proliferative neuroendocrine cells within the bronchial epithelium beyond the basement membrane, with the cell mass measuring 5 mm or greater in diameter. Carcinoid tumors are further subdivided into typical (84%–90%) and atypical (10%–16%) types. Typical carcinoid tumors demonstrate a nearly 1 : 1 male to female ratio, whereas atypical carcinoid tumors have a 2 : 1 female predominance. Although lung inflammation secondary to smoking has been correlated with atypical carcinoid development, no such association has been seen with the more common typical carcinoid tumor. Typical carcinoid tumors have a mean age of presentation of 46 years, whereas patients with atypical carcinoid tumors are typically 50 to 60 years old. Carcinoid tumors are the most common primary pulmonary neoplasm in children and adolescents.
Clinical Presentation
Centrally located carcinoid tumors usually manifest with symptoms, most commonly cough and hemoptysis. Patients present less commonly with wheeze, recurrent infection, chest pain, dyspnea, and constitutional symptoms. Peripheral tumors are usually asymptomatic and are discovered incidentally because of a CT performed for other reasons.
Despite the neuroendocrine cell origin of these neoplasms, paraneoplastic syndromes are rare. Cushing syndrome is reported in approximately 2% of bronchial carcinoids and manifests with hypokalemia and elevation of adrenocorticotropic hormone. Although carcinoid syndrome can be seen when gastrointestinal carcinoid tumors metastasize to the liver and release serotonin into the systemic blood system, this is extremely rare with pulmonary carcinoids. When patients develop carcinoid syndrome from a pulmonary primary, it is typically in those with metastatic disease to the liver. The carcinoid syndrome is characterized by skin flushing, severe diarrhea, bronchoconstriction, and right-sided valvular disease.
Pathophysiology
Typical carcinoid tumors are defined as cell masses 5 mm or greater in size that lack necrosis and demonstrate fewer than 2 mitoses per 10 high-power fields (HPFs). Atypical carcinoid tumors are diagnosed when there is cellular necrosis or 2 to 10 mitoses per 10 HPFs. Although diagnosis of these entities is usually evident on hematoxylin-eosin staining, antibodies directed to CD56 or components of the neurosecretory granules, including chromogranin and synaptophysin, can be used in equivocal cases. Carcinoid tumors are typically composed of polygonal cells with eosinophilic cytoplasm and finely granular chromatin most commonly arranged in an organoid or trabecular pattern.
No single staining method is diagnostic for differentiating typical from atypical carcinoid tumors. Differentiation of carcinoid from more aggressive neoplasms, such as small cell lung cancer, may be difficult with crushed biopsy specimens. In these cases staining with Ki-67 may be helpful as it assesses proliferation rate and is elevated in small cell lung cancer and large cell neuroendocrine carcinoma.
Imaging Findings
Radiography
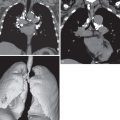
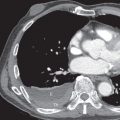
Typical and atypical carcinoids share similar imaging features. Most often this is a centrally located well-defined solitary nodule or mass with or without lobular borders in the main ( Fig. 19.4 ), lobar, or segmental bronchi. Tumors range in size from 2 to 5 cm, and calcification within the lesion is infrequently detected on radiography. They frequently cause post–obstructive atelectasis ( Fig. 19.5 ) and/or pneumonia. Segmental atelectasis or pneumonia may show periodic exacerbations and remissions, likely secondary to the ball-valve mechanism of the central lesion.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


