Transverse myelitis is an acute inflammatory condition. A relatively rare condition, the diversity of causes makes it an important diagnostic challenge. An approach to the classification and work-up standardizes diagnostic criteria and terminology to facilitate clinical research, and forms a useful tool in the clinical work-up for patients at presentation. Its pathogenesis can be grouped into four categories. Imaging appearances can be nonspecific; however, the morphology of cord involvement, enhancement pattern, and presence of coexistent abnormalities on MR imaging can provide clues as to the causes. Neuroimaging is important in identifying subgroups that may benefit from specific treatment.
Transverse myelitis can occur at any age, but is reported to have bimodal peaks in the second and fourth decades. Clinical symptoms of bilateral weakness, sensory disturbance, and autonomic dysfunction typically evolve over hours or days, most progressing to maximal clinical severity within 10 days of onset. A rostral border of sensory loss is usually thoracic, but pediatric cases have a higher rate of cervical cord involvement. At maximal clinical severity, 80% to 94% of patients have numbness, paresthesia, or band-like dysesthesia; half have a paraparesis; and almost all have bladder dysfunction to some degree.
The prognosis is highly variable; postinfectious cases in children typically have good outcomes, while other patients can be left with devastating neurologic deficits. The optimal treatment will depend upon the underlying etiology, but generally immunomodulatory treatments including steroids, immunoglobulins or plasmapheresis play a major role.
Owing to the heterogeneous pathogenesis and prognosis of transverse myelitis, it is vitally important that there be uniformly applied criteria for diagnosis and classification. This is useful, not only to guide individual patient management, but also to assess efficacy of current treatment regimes and allow identification of targets for potential novel therapies.
The diagnostic criteria devised by the Transverse Myelitis Consortium Working Group ( Box 1 ) aims to delineate transverse myelitis from the broader spectrum of transverse myelopathies and to divide transverse myelitis into idiopathic and disease-associated groups.
Inclusion criteria for diagnosis of transverse myelitis (idiopathic or disease associated)
Development of sensory, motor, or autonomic dysfunction attributable to the spinal cord
Bilateral symptoms
Clearly defined sensory level
Exclusion of compressive causes by MR imaging or CT myelography
Spinal cord inflammation demonstrated by cerebrospinal fluid pleocytosis, elevated IgG, or gadolinium enhancement
Progression to clinical nadir between 4 hours and 21 days from onset of symptoms
Exclusion criteria for diagnosis of transverse myelitis (idiopathic or disease associated)
History of irradiation to the spine within 10 years
Clear arterial distribution clinical defect consistent with anterior spinal artery occlusion
Abnormal flow voids on the surface of the cord consistent with arteriovenous malformation
Exclusion criteria for idiopathic transverse myelitis
Serologic or clinical evidence of connective tissue disease (sarcoidosis, Behçet disease, Sjögren syndrome, systemic lupus erythematosus, or mixed connective tissue disorder)
Central nervous system manifestations of syphilis, Lyme disease, HIV, HTLV-1, Mycoplasma , or other viral infection.
Brain abnormalities suggestive of multiple sclerosis
History of clinically apparent optic neuritis
The first requirement of the diagnostic criteria is an appropriate clinical picture, with symptoms referable to the cord and bilateral but not necessarily symmetric involvement. Cases with a history of spinal irradiation and those with a clear arterial distribution to their deficit are excluded. MR imaging is performed urgently to exclude compressive causes and other causes of myelopathy such as spinal arteriovenous malformation.
The second requirement of the diagnostic criteria is that evolution of symptoms to the maximal clinical severity must be between 4 hours and 21 days. The lower limit of 4 hours onset is designed to prevent cases of cord infarction, typically of abrupt onset, from being erroneously diagnosed as transverse myelitis.
The third requirement is confirmation of an active inflammatory process by presence of a cellular infiltrate and/or elevated protein on cerebrospinal fluid (CSF) analysis, or by gadolinium enhancement on MR imaging of the spinal cord.
In patients who meet the criteria for transverse myelitis, idiopathic is separated from disease-associated transverse myelitis by specifying further criteria related to presence of clinical, laboratory, or imaging evidence of connective tissue diseases, infective conditions, multiple sclerosis, and optic neuritis.
Thus, the diagnostic work-up of transverse myelitis begins with contrast-enhanced MR imaging of the spinal cord and brain, CSF analysis including cell count and protein, an autoimmune screen, and specialized tests such as serum and CSF culture, serology, and polymerase chain reaction (PCR).
A study evaluating the diagnostic criteria found that patients presenting with transverse myelopathy could be grouped as 15.6% idiopathic, 20.5% systemic disease, 17.3% infectious or parainfectious, 10.8% multiple sclerosis, and 17% neuromyelitis optica, with a further 18.8% found to have spinal cord infarct instead of transverse myelitis.
Idiopathic transverse myelitis
Idiopathic transverse myelitis made up 16.5% of acute myelopathy presentations in one large series. Heterogeneity in patient demographics, response to therapy, and outcome suggest that this group remains diverse in its pathogenesis. It is expected that better understanding of the various known causes of transverse myelitis and the development of more sensitive and specific microbial assays and autoimmune markers will cause this percent to shrink. It is yet to be determined whether there is a unified pathogenetic entity of idiopathic transverse myelitis.
Prognosis
Outcomes have been heterogeneous in this group of patients—nearly equally divided between little-to-no residual disability, moderate residual disability, and severe disability. However, the reported rates of good outcomes have ranged between 20% and 64% in different studies.
Studies have found that elevated CSF interleukin (IL)-6 levels in transverse myelitis patients correlate with and are strongly predictive of disability, suggesting that IL-6 may be the central mediator of tissue injury in transverse myelitis. Protein 14-3-3, thought to be a marker of axonal damage and a sensitive marker of Creutzfeldt-Jakob disease, has also been found in the CSF of transverse myelitis patients. Some, but not all, studies have found that this may correlate with worse outcome or progression to multiple sclerosis.
Imaging Features
MR imaging of the spinal cord
MR imaging demonstrates T2-hyperintense spinal cord lesions in almost all reported cases of idiopathic transverse myelitis. The classic lesion is predominantly central, extends over more than two segments, and involves more than two-thirds the cross-sectional area of the cord ( Fig. 1 ). The typical description includes a preference for the thoracic cord ; however, two studies have found cervical lesions are more common than thoracic (44% cervical vs 37% thoracic, and 60% cervical vs 33% thoracic, respectively).
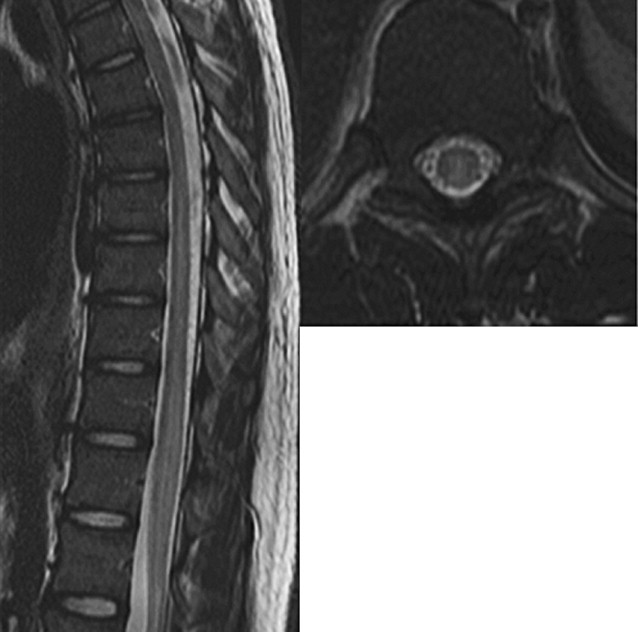
Cord expansion is described in approximately half of cases. Enhancement is present in 37% to 74% and has been reported to be more frequent in the subacute stage than at initial acute presentation. The pattern of enhancement is variable, and has been described as moderate diffuse enhancement, poorly defined heterogeneous enhancement, nodular enhancement or peripheral enhancement.
Diffusion tensor imaging
Although not currently a routine part of imaging assessment of transverse myelitis, MR diffusion tensor imaging has recently been described in transverse myelitis and, in the future, may assist in characterization of the lesion and in assessment of prognosis. A small study by Renoux and colleagues compared healthy subjects to 15 patients who had a diverse range of inflammatory myelopathies, confirming that fractional anisotropy (FA) is reduced within the cord lesion of myelitis patients. They also found that diffusion tensor imaging was more sensitive than T2-weighted imaging in identifying lesions. In 80% of patients, additional FA lesions were identified and, in 41.6% of these, the T2-occult lesion better correlated with the level of clinical manifestation than the T2-apparent lesion. A more recent study compared 10 patients with idiopathic transverse myelitis to healthy subjects. The degree of FA reduction within the lesion and presence of reduced FA in distal normal-appearing cord both correlated with worse outcomes.
Longitudinally extensive versus acute partial transverse myelitis
The length of the cord lesion, not currently specified in the diagnostic criteria, may be turn out to be an important discriminator in terms of pathogenesis and prognosis. Some researchers have differentiated between the so-called longitudinally extensive transverse myelitis (LETM), which extends over more than three segments and is centrally located involving all or most of the cross section of the cord, and acute partial myelitis, which has fewer than two segments of cord involvement and is eccentrically located or asymmetric, suggesting the prognosis may be worse in the patients with a centrally located long-segment lesion.
The morphology of the cord lesion and presence of abnormalities on the MR imaging of the brain have also been found to correlate with the risk of relapsing disease that eventually satisfies criteria for multiple sclerosis. When the cord lesion is long-segment and central in location, the risk of progression to clinically definite multiple sclerosis is low, at less than 2%. An asymmetric cord lesion less than two segments in length in combination with a normal initial MR imagining of the brain has 10% risk of progression to clinically definite multiple sclerosis over 61 months follow-up ; this increases to 21% risk of progression at 20 years follow-up according to another study of clinically isolated syndromes such as transverse myelitis and optic neuritis. When the initial MR imaging of the brain shows the presence of nonspecific white matter lesions, the risk of progression to multiple sclerosis is substantially increased, with rates of up to 88% reported.
Multiple sclerosis and neuromyelitis optica
Multiple Sclerosis
Rarely, transverse myelitis may be the initial presentation of multiple sclerosis, a multiphasic, inflammatory, demyelinating disorder affecting brain, cord, and optic nerves, with the highest incidence seen in young-adult females. Diagnosis requires clinical and MR imaging evidence of characteristic lesions demonstrating dissemination in time and space. The diagnosis is supported by the presence of oligoclonal bands in CSF, which are found in more than 80% of patients. Visually evoked response testing may reveal subclinical optic nerve involvement.
In contrast to transverse myelitis, the classic spinal cord lesions in multiple sclerosis are small, involve less than two segments, and predominantly affect the peripheral cord in a dorsal and lateral location ( Fig. 2 ). Active lesions are T2 hyperintense, with gadolinium enhancement. In the subacute to chronic phase, enhancement and swelling resolves. Old lesions may become T1 hypointense or be associated with focal atrophy. This dissemination in time is a key concept in differentiating multiple sclerosis from transverse myelitis. In the rare instances where multiple sclerosis presents as transverse myelitis, follow-up imaging will demonstrate new lesions occurring in cord and brain.
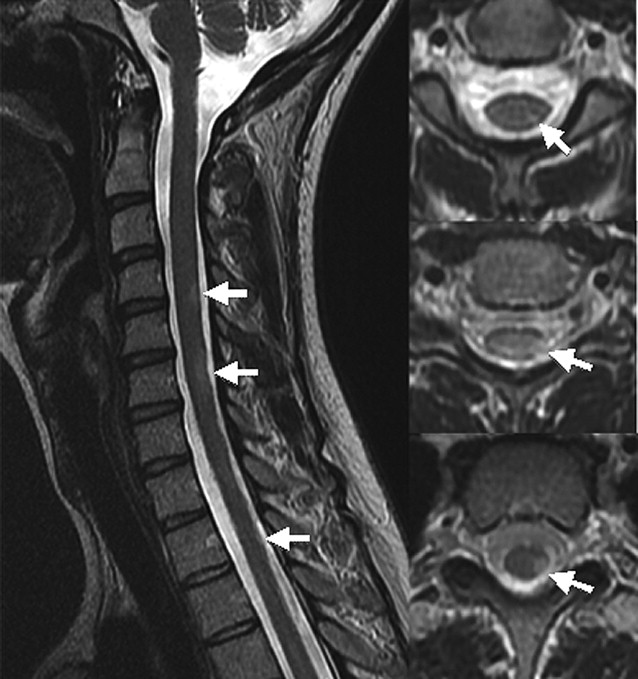
Neuromyelitis Optica
This severe relapsing demyelinating condition, previously referred to as Devic disease, affects the cord and optic nerves with relative sparing of the brain. The disease is demographically and clinically distinct from multiple sclerosis, affecting an older age group (39 years compared with 29 years), with more pronounced female predominance, and with disproportionate representation from the nonwhite population. Attacks are frequently more severe than in multiple sclerosis and lead to greater residual disability, with more than 50% of patients visually impaired and requiring ambulatory assistance at 5 years from diagnosis. CSF analysis reveals pleocytosis with high proportion of neutrophils, and oligoclonal bands are present in only 15% to 30% of neuromyelitis optica (NMO) cases compared with 85% of multiple sclerosis cases.
The concept of NMO as a condition discrete from other forms of demyelination has been strengthened by the discovery of an NMO-specific autoantibody that binds to aquaporin 4, the water channel protein most abundant in the CNS, which is highly specific (91%) and sensitive (73%) to NMO. Further refinements to the immunoassay have achieved sensitivity and specificity of 91% and 100%, and higher titers of the antibody have been demonstrated to correlate with disease severity and clinical relapse. The 2006 revised diagnostic criteria for NMO reflects these developments. The diagnostic combination of two out of three criteria provides 99% sensitivity and 90% specificity for the NMO. These include NMO IgG positivity, longitudinally extensive cord lesion, or onset MR imaging of the brain which is nondiagnostic for multiple sclerosis.
On MR imaging, the T2-hyperintense cord lesions are typically centrally located and span more than three segments. Enhancement is variable, but patchy central enhancement and expansion is common in the acute phase. The myelitis can extend cranially to involve brainstem with resultant hiccups, nausea, and respiratory failure.
MR imaging evidence of brain involvement is uncommon at presentation, but the most patients will develop nonspecific and clinically silent white matter lesions later in the course. In 10% of patients, multiple sclerosis-like cerebral lesions have been described, but were reported to have a subtly differing appearance on postgadolinium T1 sequences, described as “cloud-like” with patchy enhancement and indistinct margins. In another 10%, involvement of the hypothalamus and periaqueductal gray matter has been described. This is thought to be typical of NMO and is explained with reference to the high expression of aquaporin 4 at these sites.
Of NMO patients, 10% to 40% have a coexistent autoimmune disorder such as SLE, Sjögren syndrome or myasthenia gravis, presumably reflecting susceptibility to autoimmune conditions.
NMO IgG has become a very useful tool in the work-up of transverse myelitis. The combination of NMO-IgG positivity and LETM implies that NMO is the underlying cause. Patients can receive the optimal treatment for their myelopathy and are excluded from the idiopathic transverse myelitis cohort in research projects.
Multiple sclerosis and neuromyelitis optica
Multiple Sclerosis
Rarely, transverse myelitis may be the initial presentation of multiple sclerosis, a multiphasic, inflammatory, demyelinating disorder affecting brain, cord, and optic nerves, with the highest incidence seen in young-adult females. Diagnosis requires clinical and MR imaging evidence of characteristic lesions demonstrating dissemination in time and space. The diagnosis is supported by the presence of oligoclonal bands in CSF, which are found in more than 80% of patients. Visually evoked response testing may reveal subclinical optic nerve involvement.
In contrast to transverse myelitis, the classic spinal cord lesions in multiple sclerosis are small, involve less than two segments, and predominantly affect the peripheral cord in a dorsal and lateral location ( Fig. 2 ). Active lesions are T2 hyperintense, with gadolinium enhancement. In the subacute to chronic phase, enhancement and swelling resolves. Old lesions may become T1 hypointense or be associated with focal atrophy. This dissemination in time is a key concept in differentiating multiple sclerosis from transverse myelitis. In the rare instances where multiple sclerosis presents as transverse myelitis, follow-up imaging will demonstrate new lesions occurring in cord and brain.
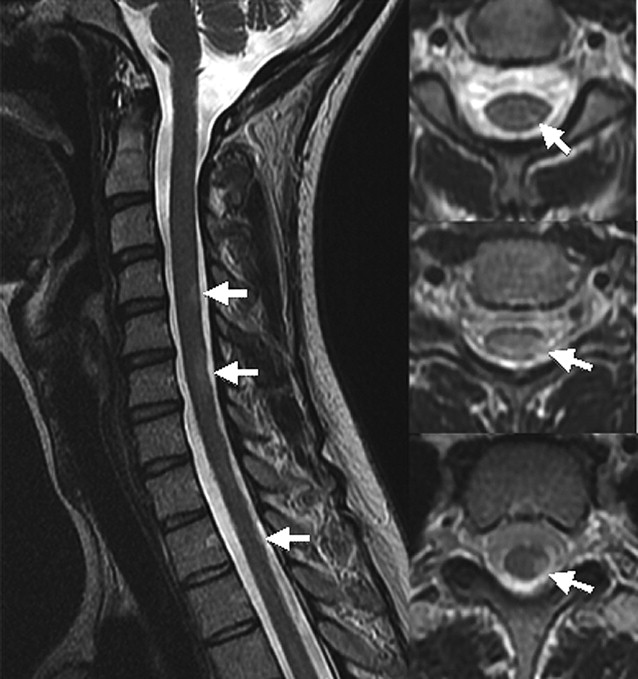
Neuromyelitis Optica
This severe relapsing demyelinating condition, previously referred to as Devic disease, affects the cord and optic nerves with relative sparing of the brain. The disease is demographically and clinically distinct from multiple sclerosis, affecting an older age group (39 years compared with 29 years), with more pronounced female predominance, and with disproportionate representation from the nonwhite population. Attacks are frequently more severe than in multiple sclerosis and lead to greater residual disability, with more than 50% of patients visually impaired and requiring ambulatory assistance at 5 years from diagnosis. CSF analysis reveals pleocytosis with high proportion of neutrophils, and oligoclonal bands are present in only 15% to 30% of neuromyelitis optica (NMO) cases compared with 85% of multiple sclerosis cases.
The concept of NMO as a condition discrete from other forms of demyelination has been strengthened by the discovery of an NMO-specific autoantibody that binds to aquaporin 4, the water channel protein most abundant in the CNS, which is highly specific (91%) and sensitive (73%) to NMO. Further refinements to the immunoassay have achieved sensitivity and specificity of 91% and 100%, and higher titers of the antibody have been demonstrated to correlate with disease severity and clinical relapse. The 2006 revised diagnostic criteria for NMO reflects these developments. The diagnostic combination of two out of three criteria provides 99% sensitivity and 90% specificity for the NMO. These include NMO IgG positivity, longitudinally extensive cord lesion, or onset MR imaging of the brain which is nondiagnostic for multiple sclerosis.
On MR imaging, the T2-hyperintense cord lesions are typically centrally located and span more than three segments. Enhancement is variable, but patchy central enhancement and expansion is common in the acute phase. The myelitis can extend cranially to involve brainstem with resultant hiccups, nausea, and respiratory failure.
MR imaging evidence of brain involvement is uncommon at presentation, but the most patients will develop nonspecific and clinically silent white matter lesions later in the course. In 10% of patients, multiple sclerosis-like cerebral lesions have been described, but were reported to have a subtly differing appearance on postgadolinium T1 sequences, described as “cloud-like” with patchy enhancement and indistinct margins. In another 10%, involvement of the hypothalamus and periaqueductal gray matter has been described. This is thought to be typical of NMO and is explained with reference to the high expression of aquaporin 4 at these sites.
Of NMO patients, 10% to 40% have a coexistent autoimmune disorder such as SLE, Sjögren syndrome or myasthenia gravis, presumably reflecting susceptibility to autoimmune conditions.
NMO IgG has become a very useful tool in the work-up of transverse myelitis. The combination of NMO-IgG positivity and LETM implies that NMO is the underlying cause. Patients can receive the optimal treatment for their myelopathy and are excluded from the idiopathic transverse myelitis cohort in research projects.
Systemic autoimmune conditions
Transverse myelitis in autoimmune conditions has most frequently been described in SLE, with and without antiphospholipid syndrome, but well-documented cases have implicated Sjögren syndrome, mixed connective tissue disease, antiphospholipid syndrome, sarcoidosis, Behçet disease, rheumatoid arthritis, and ankylosing spondylitis. Although transverse myelitis is an uncommon complication of connective tissue diseases, sufferers bear a 1000-fold greater risk than the general population. It is also important to note that transverse myelitis may be the initial presentation; in one study of transverse myelitis presentations, almost half of those eventually attributed to a systemic autoimmune disease were not aware of the diagnosis at presentation.
The pathogenesis of transverse myelitis in this population is not well understood. Scarce autopsy studies in SLE patients have described ischemic necrosis, infarction, or myelomalacia of the cord, and perivascular inflammatory cells and occasionally thrombi in small vessels. Historically, the favored hypothesis has been that vasculitis and arterial thrombi related to autoimmunologic phenomena result in ischemic necrosis within the cord.
Advances in the understanding of the pathogenesis of NMO have given rise to an alternative hypothesis. Pointing to the well-documented high incidence of optic neuritis and the more recent evidence of NMO-IgG positivity in many cases, it has been suggested that, in most cases, myelitis in patients with a connective tissue disorder may represent coexistent NMO instead of a direct complication of the autoimmune disorder.
However, more recent work on a cohort of SLE patients described two clinical pictures of myelopathy. One was a profound acute onset weakness with poor prognosis that occurred in the setting of active SLE. The other had a more subacute course of progressive weakness associated with optic neuritis and NMO positivity. A review of Sjögren-associated myelopathy cases postulated a similar division. This suggests that although coexistent NMO is an important cause of myelopathy in the setting of systemic autoimmune diseases, there is a separate entity of transverse myelitis related to the underlying vasculitic process.
SLE
CNS manifestations in SLE are common, occurring in more than half of patients and including aseptic meningitis, cerebrovascular disease, seizures, cranial and peripheral neuropathy, cognitive and psychological disorders, and headache. Myelopathy is an uncommon manifestation, with 1% to 3% incidence but, importantly, is the initial presentation in almost half of cases.
The clinical presentation of acute SLE-associated myelitis has been described as acute in onset, accompanied by a short prodrome of fever, headache, and malaise. Coexistent optic neuritis is present in 21% to 48%, and CSF protein levels are elevated without oligoclonal bands. Correlation with markers of lupus activity and autoantibodies such as antiphospholipid antibodies is variable in the reported literature.
Prognosis is variable, with complete recovery in 50%, partial recovery in 29%, and no improvement or deterioration in 21%. Worse outcomes have been described in patients with rapid onset of profound weakness at presentation, and in those with initial MR imaging findings of extensive diffuse spinal cord involvement. Conversely, rapid resolution of MR imaging of the lesions following the institution of immunosuppressive therapy suggests a favorable outcome.
MR imaging findings also vary in the reported literature. MR imaging of the spinal cord is normal at presentation in 16% to 30% of patients with clinical myelitis. Of the remainder, most of the reported cases identify long-segment central T2 hyperintensity and expansion, with enhancement common but variable; whereas isolated cases demonstrate small multifocal T2-hyperintense lesions.
Birnbaum and colleagues described two distinct subtypes of myelitis within a cohort of 22 patients with SLE and myelitis, the largest such cohort yet studied. One group had a rapid onset (<6 hours in 72.7%) of flaccid paralysis, hyporeflexia, and urinary retention, leading to a poor outcome with persistent paraplegia. The second group had a more subacute onset of spasticity, hyperreflexia, and mild-to-moderate weakness for 1 to 30 days, followed by a relapsing course.
Group 1 was differentiated by a febrile prodrome, evidence of active SLE based on clinical grounds and erythrocyte sedimentation rate (ESR), and CSF profile of neutrophilic pleocytosis and elevated protein. Incidence of optic neuritis (0% in group 1 and 54.5% in group 2) and NMO-IgG positivity (12.5% in group 1 and 57.1% in group 2) was also strikingly different between the two groups. These findings suggest both NMO and disease-associated transverse myelitis exist in SLE.
Sjögren Syndrome
The sicca complex of keratoconjunctivitis sicca, xerostomia, and salivary gland inflammation is the hallmark of this chronic progressive autoimmune-mediated inflammatory exocrinopathy, but systemic disease can affect all organ systems. The syndrome is divided into primary and secondary Sjögren syndrome, with the latter most commonly associated with rheumatoid arthritis, although other autoimmune disorders such as SLE are also associated. Women are affected more frequently than men (ratio 9:1), with onset commonly at 40 to 50 years of age.
CNS involvement has been described in 25% to 30% of patients, and includes demyelination, autonomic dysfunction, stroke, peripheral neuropathy, and myelopathy. Transverse myelitis is rare, occurring in up to 1% of patients with Sjögren syndrome, and the experience within medical literature is limited to case reports.
Williams and Butler reviewed the 17 previously reported cases and divided cases into two clinical patterns. The first is a rapid onset of severe sensory and motor deficit accompanied by neck and interscapular pain, with poor outcome and high mortality. The second is a subacute progression from gait, sensory, and urinary disturbance to eventual paraplegia, associated with optic neuritis in four out of five cases. As in SLE, NMO and disease-associated transverse myelitis both seem to be represented in this population.
Most of the cases that have undergone MR imaging demonstrate a long-segment central cord lesion with cord expansion and variable enhancement, but multifocal lesions have also been described.
Behçet Disease
The clinical triad of uveitis with recurrent oral and genital ulcers is the hallmark of Behçet disease, an episodic systemic inflammatory disease that can involve almost all tissues. The disease is most prevalent in the eastern Mediterranean, Middle East, and eastern Asia, with the highest prevalence in Turkey (80–370 cases per 100,000 population). The disease is rare in the western world (0.12–0.33 cases per 100,000 population in the United States). Age at onset is typically between 20 to 40 years, and the disease is twice as common in men as in women.
CNS manifestations arise in around 10% of patients and are usually divided into parenchymal, including meningoencephalitis (in 75%) or myelitis (in 10%), and nonparenchymal, including venous thrombosis (in 18%), idiopathic intracranial hypertension, and, rarely, intracranial aneurysm formation. CNS manifestations are reportedly twice as common in male as in female patients.
Myelitis makes up approximately 10% of cases of neuro-Behçet disease. The diagnosis in an acute transverse myelitis presentation may be suspected based on known history or presence of uveitis and ulcers, but is occasionally the presenting condition. Typically, patients have a CSF pleocytosis with normal CSF glucose. ESR correlates with active disease and is usually elevated.
MR imaging of the spinal cord shows T2-hyperintense lesions, predominantly of the posterolateral cord, which usually extend over more than two segments. Myelitis in neuro-Behçet disease has been an isolated CNS manifestation in most reports and so MR imaging of the brain is generally normal. However, some cases of spinal involvement have been identified as part of a severe case of meningoencephalitis, with the typical MR imaging findings of unilateral T2 hyperintensity, edema, and enhancement within the brainstem, thalamus, and basal ganglia. In these cases, brainstem involvement will tend to dominate the clinical picture.
Sarcoidosis
Sarcoidosis is a noncaseating granulomatous disease of uncertain cause. The African American population has a higher incidence (35–80 per 100,000) and younger onset (30–50 years) compared with white Americans (3–10 per 100 000 and onset of 40–60 years). Incidence also increases in the lower latitudes (15–20 per 100,000 in northern Europe compared with 1–5 per 100,000 in southern Europe). Women have an approximately 30% greater risk.
CNS involvement in sarcoidosis occurs in 5% to 10% of patients and, in half of these, it will be part of the presenting illness. Spinal involvement is considered relatively uncommon, estimated to occur in 6% to 8% of patients with neurosarcoidosis. However, this may be an underestimation because cases are increasingly reported in newer studies using MR imaging criteria. Indeed, Spencer and colleagues found that spinal involvement was the most common manifestation in their cohort of 21 patients with neurosarcoidosis, occurring in 43%.
The most common clinically apparent abnormalities described on MR imaging are intramedullary lesions with patchy enhancement extending more than three levels with a preference for cervical segments, although nonenhancing lesions and multifocal short-segment lesions have also been described. Associated leptomeningeal enhancement is a common finding, whereas extradural mass lesions and multiple cauda equine lesions have less commonly been described.
Parainfectious transverse myelitis
Parainfectious myelitis is the term used for transverse myelitis that is temporally related to and presumed caused by a systemic infection, although in many cases the infective agent cannot be identified. Up to 40% of cases of pediatric transverse myelitis are preceded by clinical signs or serologic evidence of a systemic infection, with viral infections most commonly implicated. In the adult population, parainfectious causes have been implicated in 6% to 45% of presentations of acute transverse myelitis.
In practice, cases of transverse myelitis related to direct infection are difficult to delineate from those occurring in the convalescent phase; therefore the term covers those cases due to direct microbial infection with neuronal injury resulting from either the infection itself or from the systemic immune process against the agent, as well as those cases occurring in the convalescent phase due to an immune-mediated response to a recent infection. The diagnosis can be made in clinical presentations of transverse myelitis with a history of an infectious syndrome within 4 weeks preceding clinical onset, accompanied by culture, serologic, or PCR evidence of infection.
MR imaging features are often nonspecific, mirroring those in idiopathic transverse myelitis. Table 1 lists some of the more commonly implicated viral, bacterial, and fungal pathogens, with a description of common imaging findings. The most common and least specific finding is of long-segment T2 hyperintensity within the cord, usually with some degree of edema. Enhancement may be diffuse, patchy, peripheral, or absent. Associated leptomeningeal and/or nerve root enhancement has been described in infection with some pathogens, including herpes simplex virus (HSV), Epstein-Barr virus, varicella-zoster virus (VZV), mycobacterium tuberculosis, Lyme disease, and parasitic agents ( Figs. 3 and 4 ).
| Clinical Features | Imaging Features on MR Imaging Spinal Cord | |
|---|---|---|
| Herpes Viruses | ||
| Herpes Simplex Virus 1 | Most common cause of viral encephalitis. Myelitis is rare and may occur as part of meningoencephalomyelitis | Long-segment ↑T2 signal with variable enhancement Case reports of intramedullary hemorrhage |
| Herpes Simplex Virus 2 | Ascending myelitis is more common than in Herpes simplex virus 1 infection | Long-segment ↑T2 signal with variable enhancement |
| Human Herpesvirus 6 and 7 | Encephalitis and meningoencephalitis are rare. Myelitis is extremely rare | Isolated case reports have showed normal MR imaging or long-segment ↑T2 signal |
| Varicella-Zoster Virus | Primary disease: cerebellar ataxia, meningoencephalitis, transverse myelitis, and aseptic meningitis occur in <1% Reactivation: radiculomyelitis is accompanied by dermatomal rash in only half of cases HIV/AIDS: disseminated disease with rash and extensive cord involvement | Long-segment ↑T2 signal centered at level of dermatome Enhancement is common Changes are most severe at dorsal root entry zone and posterior horn of the involved dermatome |
| Cytomegalovirus | CNS disease is rare in the immunocompetent Manifestations include radiculitis/radiculomyelitis (most common), myelitis, encephalomyelitis, and ventriculoencephalitis | Thickening, clumping, and enhancement of nerve roots and leptomeninges, often with associated long-segment cord ↑T2 signal, favoring conus Case reports of isolated long-segment cord ↑T2 signal and expansion |
| Epstein Barr Virus | Symptomatic CNS involvement occurs in 5%, including meningitis, encephalitis, myelitis, cranial neuritis, ADEM, and GBS | ↑T2 signal and enhancement is usually long-segment and may be multifocal Nerve root and/or leptomeningeal enhancement may be present |
| Orthomyxoviruses | ||
| Influenza A | Encephalitis is rare, and myelitis very rare | Isolated case reports of ↑T2 signal and enhancement |
| Paramyxoviruses | ||
| Measles Virus | Encephalitis and encephalomyelitis are rare manifestations | Isolated case reports describe long-segment ↑T2 signal and expansion with variable enhancement |
| Mumps Virus | Meningoencephalitis is a rare manifestation and myelitis is very rare | Long-segment ↑T2 signal and expansion |
| Picornaviruses | ||
| Poliovirus | Symptomatic CNS involvement in 1%–2%, manifesting as spinal poliomyelitis (in 75%), bulbar poliomyelitis, meningitis, or polioencephalitis | Few MR imaging studies, showing ↑T2 signal of the anterior horns |
| Enterovirus 71 | CNS involvement in up to 30%, manifested by meningitis, encephalitis, and poliomyelitis-like syndrome. Rarely transverse myelitis and GBS |
|
| Coxsackie Virus A and B | Rare cases of poliomyelitis-like syndrome and transverse myelitis. | |
| Echovirus | Rare cases of poliomyelitis-like syndrome and transverse myelitis | |
| Flaviviruses | ||
| West Nile virus | CNS complications are common and may manifest as rhombencephalitis (50%), aseptic meningitis (20%), or poliomyelitis-like syndrome (5%–10%) |
|
| Tick-Borne Encephalitis Virus | Meningitis (49%), meningoencephalitis (41%), meningoencephalomyelitis (10%), poliomyelitis-like syndrome | |
| Japanese Encephalitis Virus | Encephalitis is the most common CNS manifestation, with poliomyelitis-like syndrome occurring less commonly than in West Nile virus | |
| Murray Valley Encephalitis Virus | ||
| St Louis Encephalitis Virus | ||
| Dengue Virus | CNS involvement is uncommon, but may manifest as encephalitis, myelitis, ADEM, or GBS | |
| Retroviruses | ||
| HTLV 1 | <5% of carriers develop symptomatic neurologic involvement | Long-segment ↑T2 signal of lateral columns, less commonly extending to dorsal columns, occasionally with enhancement. Progresses to atrophy of lateral columns with no signal change or enhancement |
| HIV | HIV myelitis: isolated or with cerebral involvement Vacuolar myelopathy: present in up to 50% AIDS patients, but symptomatic in 5%–10%. Likely a metabolic complication rather than due to direct infection | HIV myelitis: Long-segment ↑T2 signal with multifocal enhancement Vacuolar myelopathy: MR imaging normal or dorsolateral ↑T2 signal over multiple segments |
| Bacterial | ||
| Borrelia burgdorferi | CNS involvement in 20% of untreated cases. Meningitis, meningoencephalitis, meningoencephalomyelitis, cranial nerve palsies, and painful radiculopathy. Meningitis and meningoencephalitis are more common in European than North American strains | Nerve root enhancement plus cord ↑T2 signal of adjacent segments. Leptomeningeal or nerve root enhancement may be present Rarely transverse myelitis picture with long-segment diffuse ↑T2 signal with patchy enhancement |
| Treponema pallidum | Neurosyphilis in 5%–10% of untreated cases. Meningitis, meningovascular disease (endarteritis), myelitis, meningomyelitis, tabes dorsalis | Short-segment peripheral and long-segment ↑T2 signal with enhancement have both been described. Leptomeningeal enhancement may be seen |
| Mycoplasma pneumoniae | Encephalomyelitis is common in children. Myelitis is a rare complication | Long-segment ↑T2 signal |
| Mycobacterium tuberculosis | CNS involvement may include meningitis, tuberculoma or, less commonly, myelitis | 80% involve thoracic cord, majority with ↑T2 signal, but may have ↓T2 signal lesions Diffuse, nodular, or ring-like enhancement Cavitation may occur Leptomeningeal and dural enhancement may be present |
| Parasites | ||
| Neurocysticercosis | Intracranial involvement is most common, with spinal involvement occurring in 1%–5% Intramedullary lesions are far less common than extramedullary intradural lesions | Intramedullary: cystic lesion that may develop irregular peripheral enhancement and edema as it degenerates Extramedullary: clusters of cystic lesions, or thick irregular leptomeningeal enhancement |
| Schistosomiasis | Spinal involvement is uncommon | ↑T2 signal and mild-to-moderate expansion of the distal cord and conus. Enhancement patterns may include nodular intramedullary foci, leptomeningeal enhancement, and enhancing thickening of nerve roots |
| Gnathostomiasis | Organism endemic in Southeast Asia Eosinophilic meningitis, myelitis, and painful radiculitis May present with subarachnoid hemorrhage | ↑T2 signal long-segment expanded lesions of the central cord. Enhancement and hemorrhage are common. Scattered intracranial lesions may coexist |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


