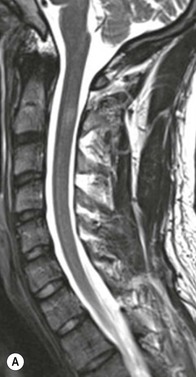
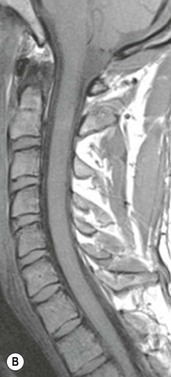
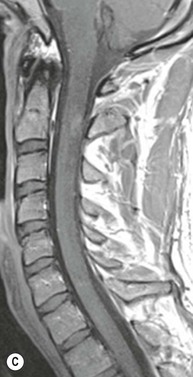
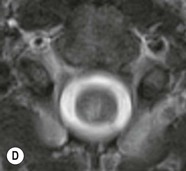
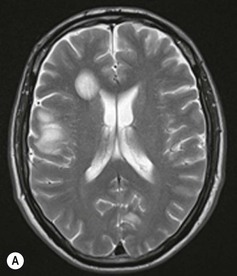
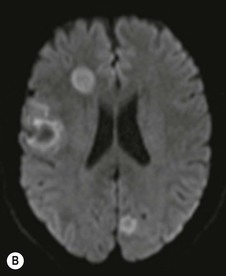
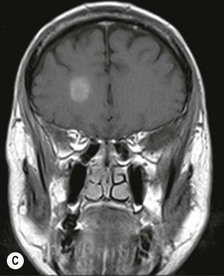
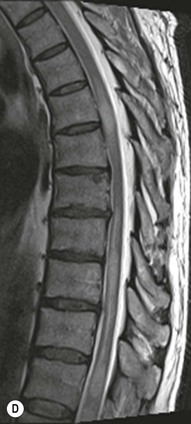
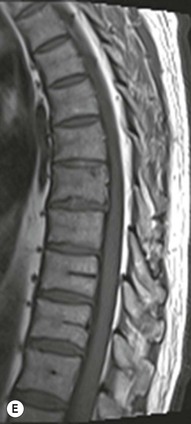
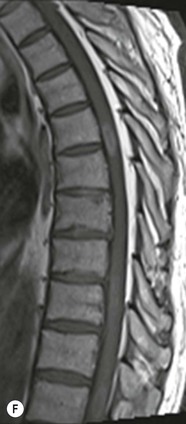
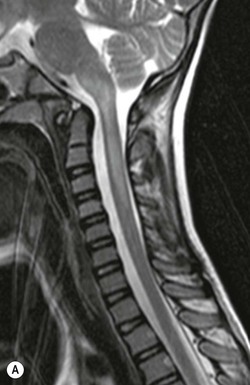
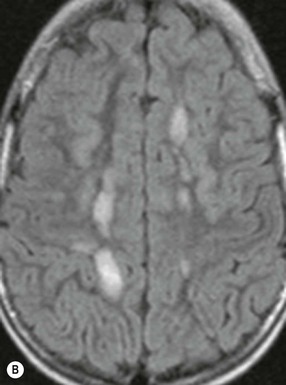
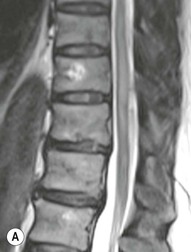
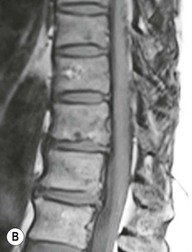
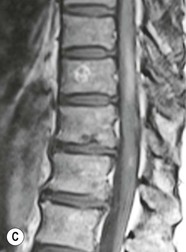
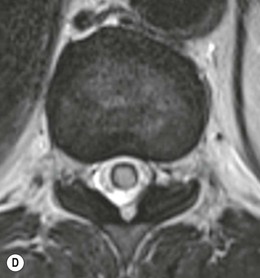
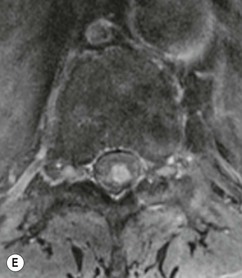
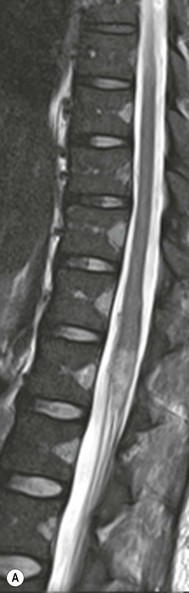
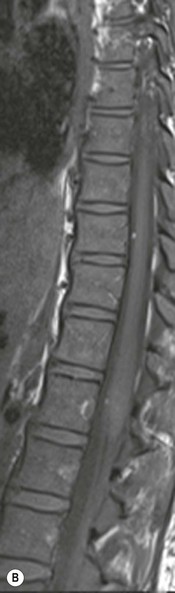
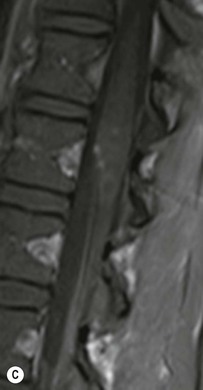
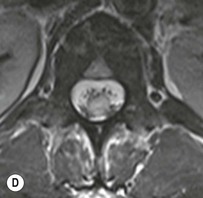
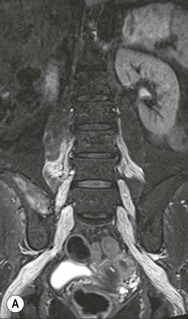
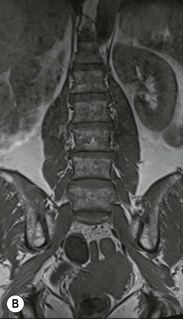
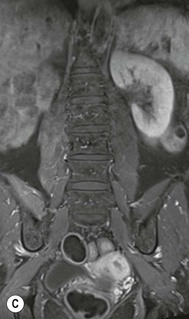
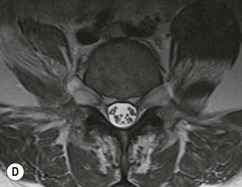
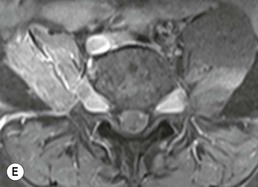
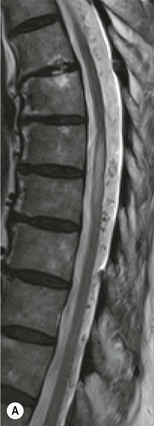
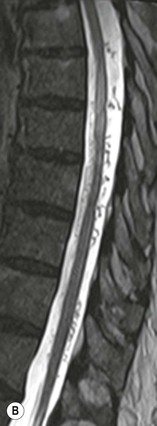
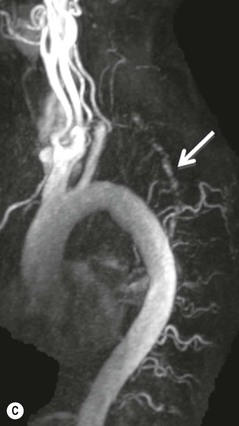
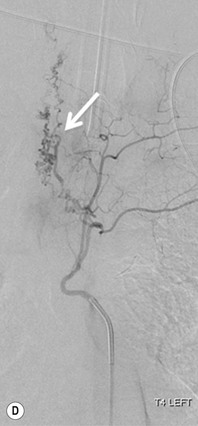
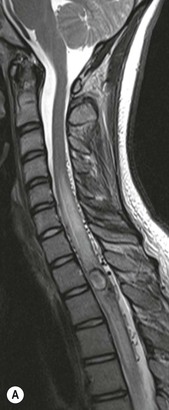
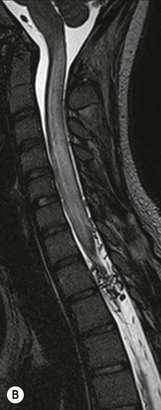
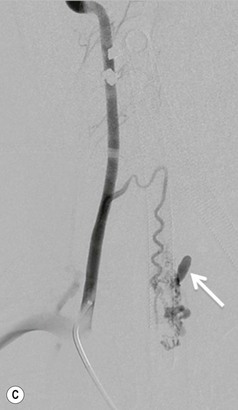
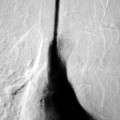
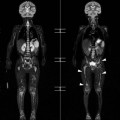
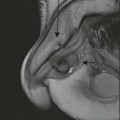
Farah Alobeidi, Majda M. Thurnher, H. Rolf Jäger Multiple sclerosis (MS) is a progressive neurodegenerative disorder characterised by multiple inflammatory demyelinating foci called ‘plaques’. The spinal cord is commonly involved with changes on autopsy in up to 98% of the cases. One-third of MS patients will have isolated spinal cord involvement. Spinal cord abnormalities in MS include focal lesions, diffuse involvement, axonal loss and spinal cord atrophy. Focal MS lesions appear as oval- or wedge-shaped T2 hyperintensities located preferentially in the lateral and posterior parts of the spinal cord, which may or may not be swollen. Lesion enhancement is seen less frequently than in the brain, and is commonly subtle (Fig. 57-1). Ring-like or intense nodular enhancement may also occur. Diffuse signal intensity abnormalities extending over multiple vertebral segments resembling transverse myelitis are seen in primary and secondary progressive MS. Spinal cord atrophy is associated with clinical disability, and is more common in the upper part of the spinal cord. Tumefactive MS lesions can sometimes present a diagnostic challenge with a clinical presentation and imaging features mimicking tumours. Magnetic resonance imaging (MRI) appearances are classically of large (greater than 2 cm) circumscribed lesions with little mass effect or oedema. They are typically found in the supratentorial white matter but can also involve grey matter and the spinal cord (Fig. 57-2). Approximately half of tumefactive lesions enhance with a typical open ring pattern, with the incomplete portion of the ring on the grey matter side of the lesion (Fig. 57-2). Corticosteroid therapy leads to a dramatic reduction in the size of the lesions. Acute disseminated encephalomyelitis (ADEM) is an inflammatory demyelinating cental nervous system (CNS) disease of the brain and spinal cord, with a distinct tendency to a perivenous localisation of pathological changes. ADEM develops mostly one or two weeks following a viral disease or prior vaccinations. Cerebrospinal fluid (CSF) analysis shows a high protein level. A high serum titre of IgG specific for myelin oligodendrocyte glycoprotein (MOG) has been described in almost one-half of the studied cases of ADEM.1 The spinal cord is involved in 30–40% of the cases. On MR imaging, non-enhancing hyperintense lesions are seen in the spinal cord on long TR sequences (Fig. 57-3). Skip lesions, as well as long segment hyperintensity, may be detected. Complete resolution of abnormalities will usually be seen on follow-up images. Acute transverse myelitis (ATM) is an aetiologically heterogeneous syndrome with acute or subacute onset, manifesting as weakness, sensory loss and autonomic dysfunction. It is associated with infectious or systemic autoimmune diseases, but in the majority of cases the aetiology remains unknown (idiopathic). In 2002 diagnostic criteria for idiopathic and disease-associated ATM were proposed by the Transverse Myelitis Consortium Working Group.2 The outcome of ATM ranges from full recovery to complete inability to walk or even death from respiratory failure. On MR imaging, intramedullary T2 high signal intensity with cord swelling will be seen. Enhancement may be present. In comparison with spinal cord involvement in MS where focal lesion do not take more than half of the cross-sectional area of the cord, lesions in TM tend to involve more than two-thirds of the cross-sectional area of the cord (Fig. 57-4). Depending on the length of the signal abnormality, TM can be divided into longitudinally extensive TM (LETM) when signal abnormalities extend more than two segments and acute partial TM (APTM) when signal abnormalities extend less than two vertebral segments. Neuromyelitis optica (NMO) is a severe inflammatory disorder that predominantly affects the optic nerves and the spinal cord. It has a relapsing course in 80% of the cases and females are more commonly affected (9 : 1). The discovery of aquaporin-4 antibodies in 2004 has substantially changed our understanding of the disease, which was for many years debated and considered as an MS subtype.3 Antibodies to aquaporin-4 (AQP4-Ab or NMO-IgG) are sensitive and highly specific serum markers of NMO, and will be positive in 60–90% of NMO patients. Patients with NMO present either with optic neuritis (unilateral or bilateral) or with LETM and spinal symptoms. In optic neuritis MRI shows hyperintensity of the optic nerves with enhancement. Spinal cord involvement manifests itself as intramedullary T2 hyperintense signal often extending more than three vertebral segments (LETM) with cavitations and patchy enhancement. On follow-up magnetic resonance (MR) defects, atrophy and central cavities, predominately located in the area of the posterior fascicle, have been described.4 In some cases MRI findings may resemble spinal cord ischaemia with bilateral ventral hyperintensities.4 Contrary to the common belief that brain lesions are not present in NMO, recent studies reported brain abnormalities in up to 40–60% of NMO patients. Periventricular signal intensity abnormalities (around the third and fourth ventricle, and in the periaqueductal region) can be detected on MRI, corresponding to brain areas with the highest aquaporin concentrations. Extensive lesions in the cerebral hemispheres have also been described.5 Enhancement on brain MRI is not common (13–36%), with ‘cloud-like enhancement’, which appears as multiple patches of enhancing lesions being the most common type.6 NMO has been associated with other autoimmune diseases, including hypothyroidism, Sjögren’s syndrome (SS), systemic lupus erythematosus (SLE), pernicious anaemia, ulcerative colitis, primary sclerosing cholangitis, rheumatoid arthritis, mixed connective tissue disorders and idiopathic thrombocytopenic purpura. Systemic lupus erythematosus (SLE) is a relapsing and remitting, chronic, multisystem autoimmune disease. Although the frequency of neuropsychiatric lupus has been reported as high as 95%, SLE-related myelitis is rare, with prevalence varying between 1 and 2%. Involvement of the spinal cord in SLE usually occurs during a time of acute exacerbation and is occasionally the first manifestation of SLE in an undiagnosed patient. SLE myelitis manifests mostly as transverse myelopathy. The pathophysiological mechanism of TM in SLE is uncertain, although vasculitis and arterial thrombosis resulting in ischaemic cord necrosis have been suggested. Studies have suggested a higher incidence of antiphospholipid and NMO-IgG antibodies in those with SLE myelitis than in the general SLE population and this has contributed to our understanding of the disease process.7 The mid-thoracic cord is most commonly affected, resulting in a sensory level and frequently in paraplegia, which may be complete. Cervical myelopathy and cauda equina involvement, on the other hand, often cause only partial motor and sensory loss. MRI demonstrates T2 hyperintensity and oedema, frequently with spinal cord expansion. Lesions may demonstrate patchy enhancement during the acute phase (Fig. 57-5). Improvement or resolution of these findings correlates with clinical improvement. Indeed, some patients may have a normal MRI if they have already received treatment. Sarcoidosis is a systemic condition of unknown aetiology characterised histologically by non-caseating granulomatosis. Although CNS sarcoidosis is found in approximately 25% of cases on post-mortem examination, symptomatic involvement in life is uncommon. Clinical presentation depends on the site of involvement and is often non-specific. Spinal involvement may be osseous, discal, meningeal or involve the cord itself. The most common spinal cord manifestation is leptomeningeal enhancement. This is best seen on sagittal contrast-enhanced T1 images as thin linear or nodular enhancement, which frequently extends along the surface of the nerve roots. Dural involvement is more nodular in appearance, often with enhancing dural-based mass-like lesions that may mimic meningiomas. Intramedullary spinal lesions are uncommon and frequently associated with severe neurological deficit. MRI demonstrates an enhancing mass that is hyperintense on T2 sequences with associated fusiform enlargement of the spinal cord. These findings are, however, non-specific and can mimic intramedullary tumours, TM, MS and fungal infections. When the cauda equina is involved there is enhancement and clumping of the nerve roots. The diagnosis of CNS sarcoidosis can represent a challenge on spinal cord imaging alone and is supported by the presence of typical appearances of brain sarcoidosis, such as involvement of the hypothalamic–pituitary axis, leptomeningeal enhancement or dural masses, as well as other diagnostic tests, such as elevated serum ACE levels. Although not strictly diseases of the spinal cord, demyelinating polyneuropathies merit a brief mention here as they can affect the cauda equina and other intradural nerves, leading to abnormal findings on an MRI of the spine. Guillain–Barré is an acute immune-mediated polyneuropathy. Affected individuals have a typical areflexia and ascending paralysis type of symmetrical weakness, with or without sensory loss, that starts in the feet and hands and progressively moves up the limbs to the trunk over a few days. The trigger is frequently a viral illness with the production of antibodies that cross-react with myelin in the peripheral nervous system.8 Approximately 80% make complete or near-complete recovery over a few weeks from onset, with approximately 10% developing persistent symptoms that may have a relapsing and remitting course. In approximately 10% of cases, it can be life threatening if the respiratory muscles are affected. Diagnosis is usually clinical and supplemented by nerve conduction studies and CSF examination. MRI demonstrates nerve thickening and enhancement, which is non-specific and is seen in other inflammatory disorders but is a useful diagnostic adjunct. This is an immune-mediated chronically progressive or relapsing symmetric sensorimotor disorder. It can be considered the chronic equivalent of acute inflammatory demyelinating polyneuropathy, the most common form of Guillain–Barré syndrome. In contrast to Guillain–Barré syndrome, CIDP has an insidious onset and evolves in either a slowly progressive or a relapsing manner. Preceding infection is infrequent. As with Guillain–Barré syndrome, the mainstay of treatment is immunosuppressive or immunomodulatory intervention. MRI findings are similar, with enhancing thickened nerve roots. Acute and subacute muscle denervation is demonstrated by hyperintensity within the affected muscle on fluid-sensitive sequences, such as T2-weighted or STIR images. In chronic denervation, muscle atrophy and fatty infiltration demonstrate high signal changes on T1-weighted sequences in association with volume loss. CIDP may therefore show both these changes depending on the stage of imaging (Fig. 57-6). Over 80% of spinal arteriovenous malformations represent SDAVFs located in the spinal dural mater, usually close to a root sleeve.9 Such fistulae are most commonly located in the thoracolumbar region but can occur at any level, though in the cervical spine only around the foramen magnum except in extremely rare cases. The fistula is usually supplied by one or two branches of a nearby radiculomeningeal artery and shunts often via a single vein into intradural radicular veins. The increase in spinal venous pressure results in slow venous drainage and stagnation because radicular veins and intramedullary veins share a common venous outflow. This results in the clinical findings of progressive myelopathy. SDAVFs are more common in middle-aged to elderly men and the typical presentation is of a slowly progressive gait disturbance, difficulty climbing stairs and paraesthesia. Bowel and bladder incontinence, erectile dysfunction and urinary retention are often seen late in the course of the disease but may be the presenting feature. Diagnosis is often made by radiologists, based on typical MRI imaging features that guide definitive diagnosis by spinal DSA. T2 sequences show an ill-defined central intramedullary hyperintensity extending over multiple levels with associated cord expansion. There may be a hypointense rim, most likely due to deoxygenated blood within dilated capillary vessels surrounding the congested oedema.10 Engorged perimedullary veins can be seen as flow voids, which are more pronounced on the dorsal surface compared to the ventral surface. Although these are readily depicted on standard T2 sequences, they become much more conspicuous on heavily T2-weighted sequences (constructive interference in steady state (CISS), fast imaging employing steady-state acquisition (FIESTA) or 3D turbo spin-echo (3D TSE)) (Fig. 57-7). There may be diffuse post-contrast enhancement of the spinal cord, reflecting breakdown of the blood–brain barrier. The coiled perimedullary veins are easily depicted following contrast enhancement. It should be noted that normal pial veins can be prominent at the level of the lumbar enlargement of the spinal cord and that the distribution of abnormally enlarged veins is a poor guide to the location of a dural fistula. The site of the fistula can, however, be sometimes shown by dynamic contrast-enhanced magnetic resonance angiography (MRA). Multiplanar reformats of 4D dynamic MRA sequences, such as time-resolved angiography with interleaved stochastic trajectories (TWIST), can show non-invasively the site of the SDAVF in cases where the diagnosis is uncertain and can guide digital subtraction angiography (DSA), helping to avoid unnecessary superselective injections of all possible arterial feeders (Fig. 57-7).11 These lesions are an absolute indication for DSA. When searching for a dural fistula, imaging should be slow, say one frame rate every 2 s due to delayed venous return. A large number of arteries may have to be injected before the lesion is found. The study should not be regarded as negative unless: (A) all the spinal arteries from the foramen magnum to the coccyx have been opacified adequately, or (B) the veins thought to be abnormal have been opacified and shown to drain normally.12 If a lesion is found, adjacent levels should also be injected and the major radiculomedullary arteries supplying the region must be identified. Treatment aims to stop disease progression, with prognosis dependent on the symptoms and stage of disease pre-treatment. Embolisation of arteries supplying a dural fistula may be feasible, provided the vessel to be embolised can also be shown not to supply the spinal cord. The aim of treatment is to exclude the shunting zone (i.e. the most distal part of the artery together with the most proximal part of the draining vein).13 Of course this is not always possible and a more proximal occlusion will lead to a transient improvement of symptoms, although fistula recurrence is high in these cases. Early surgical intervention should be considered for incomplete embolisations, as delay of secondary intervention is associated with a poor outcome.14 In contrast to SDAVFs, spinal arteriovenous malformations (AVMs) are fed by spinal arteries, either radicullomedullary and/or radicullopial, located in an intra- or perimedullary location, respectively. There are two distinct subtypes: (A) glomerular AVMs (also known as plexiform or nidus type), the most common, contain a cluster or nidus of abnormal vessels between the feeding artery and draining vein and (B) fistulous AVMs (also known as AVM of the perimedullary fistula type or intradural AV fistula) which are direct AV shunts commonly located superficially on the cord.15 Drainage is via spinal cord veins or perimedullary veins. In contrast to SDAVFs, these lesions are more prone to haemorrhage, a useful discriminating feature on MRI. Depending on the AVM location, haemorrhage may be intramedullary or subarachnoid. Lesions can also cause symptoms via venous congestion in a pathophysiology mechanism similar to SDAVF. The dilated intra- and/or perimedullary veins are demonstrated on MRI as serpiginous flow voids on T2 sequences, which enhance post-contrast administration. If there is venous congestion, oedema is present, seen as ill-defined intramedullary T2 hyperintensity with cord expansion. If haemorrhage is present, various intramedullary signal intensities are seen, depending on the age of the blood. A subarachnoid haemorrhage may be present. There may also be an intranidal aneurysm (Fig. 57-8). Embolisation of AVMs can sometimes be therapeutic but frequently it is palliative or ‘targeted’ to eliminate false aneurysms or to reduce hypertension or as a precursor to surgical treatment. A summary of SDAVFs and spinal AVMs, together with other types of vascular malformations and fistulas, is shown in Table 57-1.16 TABLE 57-1
Non-Tumoural Spinal Cord Lesions
Inflammatory Disease
Multiple Sclerosis
Acute Disseminated Encephalomyelitis
Acute Transverse Myelitis
Neuromyelitis Optica
Systemic Lupus Erythematosus
Sarcoidosis
Demyelinating Polyneuropathies
Guillain–Barré Syndrome
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
Vascular Diseases
Spinal Dural Arteriovenous Fistula (SDAVF)
Spinal Arteriovenous Malformations (SAVMs)
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Non-Tumoural Spinal Cord Lesions
Chapter 57