Target
Related disease
Tracers (human application)
Binding mechanism
TSPO
MS, AD, stroke, PD, HD, schizophrenia
[11C]PK11195, DAA, and PBR derivatives
Antagonist
GABA
Stroke
[11C]Flumazenil
Antagonist
Dopaminergic system
PD, HD, tardive dyskinesia, schizophrenia, autism, ADHD, drug abuse, depression
[18F]FDOPA, [11C]SCH23390
Vesicular storage
[11C]Raclopride
Dopamine transporter
[11C]PHNO
D1 antagonist
[11C]-Beta-CIT, PE2I
D2 agonist and antagonist
ß-Amyloid
AD, MCI
[11C]PIB
Staining agent
[18F]Florbetaben, etc.
NMDA
Schizophrenia
[11C]GSK-931145
Antagonist
P-Glycoprotein
Neurodegeneration
[11C]Verapamil, [11C]dLop
Substrate
Cholinergic system
AD, PD, HD, schizophrenia
[11C]MP4A
Acetylcholinesterase inhibitor
[18F]TZTP
M2 antagonist
mGlu-5
Depression, anxiety, schizophrenia, PD
[11C]ABP688
antagonist
VMAT
PD, AD, HD
[11C]DTPZ
Adenosine receptor
PD, AD, epilepsy, sleep, neuroinflammation
[11C]MPDX
A1 antagonist
[11C]TSMX
A2a antagonist
Serotonergic system
Depression, anxiety, OCD, schizophrenia
[11C]DASB
Serotonin transporter
[11C]WAY100635
5-HT1A antagonist
[11C]MDL100907
5-HT2A antagonist
Norephedrine
Depression
[11C]Methylreboxetine
NE transporter
Opioid receptors
Analgesia, shock, appetite, thermoregulation
[11C]Diprenorphine
Kappa receptor antagonist
Monoamine oxidase
Neurodegenerative and inflammatory processes
[11C]Deprenyl
MAO inhibitors
[11C]Harmine
Energy
Neurodegenerative and inflammatory processes
[18F]FDG
Glucose consumption
Neuronal activity
Neurodegenerative and inflammatory processes
[15O]Water
Blood flow
An ideal nuclear medicine imaging tracer for brain imaging should fulfill the following criteria:
Simple automated synthesis procedure suitable for reliable production and low radiation burden for personnel. For clinical studies, GMP compliance is a prerequisite.
Appropriate specific activity which should be sufficiently high that binding of the tracers is minimally affected by nonradioactive counterparts. Especially for targets with low densities, the high specific activity is an important issue.
LogP between 1.5 and 4 in order to passively penetrate the lipophilic blood–brain barrier enabling high accumulation of target bound radioactivity.
High affinity (expressed as B max/K D) to achieve sufficient radioactive signal bound at the target and high specificity so that measured radioactive signal represents binding to the target of interest.
Metabolic stability to ensure that measured radioactivity represents binding of the administered tracer and not the metabolite.
Low affinity for P-glycoprotein since P-glycoprotein can transport tracers out of the brain resulting in low brain uptake.
Appropriate radionuclide so that the radioactive half-life is in agreement with the rate of the physiological process of interest.
So to summarize, its radioactivity accumulation should represent target density or functionality enabling the possibility to obtain quantitative data (Elsinga 2002). Since the ideal tracer does not exist, interpretation of PET data should be handled with care. In this chapter, different neurological tracers are described ranked by the molecular target.
1.2 Glucose Consumption
The most general applied tracer in PET and for neurology in particular is 2-[18F]fluoro-2-desoxyglucose (FDG). Using FDG regional cerebral glucose consumption rate can be measured quantitatively. In several neurological diseases (dementia, PD, AD, stroke), glucose consumption is reduced in specific brain regions indicating impaired functionality of these areas. Numerous articles including review papers on the use of FDG have appeared in the literature in many cases applied in combination with targeted tracers as described in this chapter (Demetriades 2002; Mielke and Heis 1998). In addition, several chapters in this volume will address the use of FDG-PET imaging.
1.3 Translocator Protein TSPO (Formerly Named Peripheral Benzodiazepine Receptor)
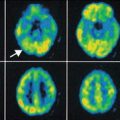
Microglia are cells in the brain governing the activity of other immune cells. Activated microglia cells are correlated with neuroinflammation, which plays an important role in the onset of neurodegenerative disease. TSPO, formerly known as the peripheral benzodiazepine receptor (PBR), is overexpressed at activated microglia (Politis et al. 2012). Several PET tracers binding to TSPO have been developed. (R)-[11C]PK11195 [1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide] is the first non-benzodiazepine and selective TSPO PET ligand (Hashimoto et al. 1989). The compound has nanomolar affinity for TSPO (Chauveau et al. 2008). The PET tracer is widely applied in TSPO PET imaging. (R)-[11C]PK11195 has been used in many human CNS studies, including multiple sclerosis, Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis, Huntington’s disease, HIV, herpes encephalitis, and schizophrenia. Although (R)-[11C]PK11195 shows increased brain uptake in several neurodegenerative disorders, there are several disadvantages. The sensitivity is low and it is difficult to quantify small changes in TSPO expression in vivo. (R)-[11C]PK11195 displays high plasma protein binding and high nonspecific binding because of its high lipophilicity. The lack of sensitivity and specificity of (R)-[11C]PK11195 has hampered development of a standard quantitative method of analysis. For this reason, several new TSPO tracers have been developed. Most of these new TSPO tracers are still in the early stage of investigation. Many research efforts in preclinical studies are aimed at the following issues: (1) metabolic stability of the tracer, (2) binding potential (BP), (3) ligand receptor kinetics, and (4) quantitation methods. R-[11C]PK11195 PET has been used mainly for assessing microglia/macrophage activation as an indication of neuronal and tissue damage in various neurodegenerative disorders. Most of these human studies used a reference tissue to measure the uptake of radioactivity. A kinetic plasma (metabolite-corrected) input with a reversible two-tissue compartment model was shown to be the best for analyzing R-[11C]PK11195 kinetics in brain.
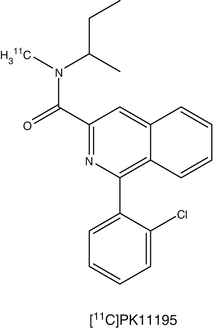
Radioligands such as [11C]PBR28, [11C]DAA1106, [18F]FEDAA1106, and [18F]PBR111 have been developed to image TSPO in vivo (Dollé et al. 2009; de Vries et al. 2006). Published data using these second-generation PET tracers [11C]DAA1106 and [18F]FEDAA1106 in humans are promising, as they showed significantly higher brain uptake than [11C]PK11195. Furthermore, increased [11C]DAA1106 binding was reported in AD patients. A study using [11C]PBR28 found areas of focal increases in radiotracer binding in the brain of multiple sclerosis patients.
1.4 GABA Receptor
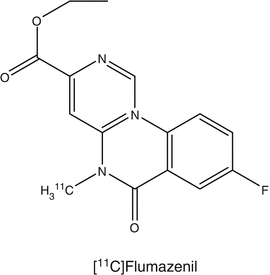
The development of epilepsy has been associated with the impairment of gamma-aminobutyric acid (GABA) neurotransmission in the brain. The high-affinity ligand [11C]flumazenil has been used widely for the investigation of GABA receptors (Hammers 2004) and was labeled in various positions (Halldin et al. 1988). [11C]Flumazenil is available commercially in the United States and has been approved by Federal Drug Agency (FDA) for use in various clinical trials. Using [11C]flumazenil with PET in patients with acute hemispheric ischemic stroke, it was shown that this tracer is a suitable tracer to distinguish between irreversibly damaged and viable neuronal tissue during early onset of the disease. With [11C]flumazenil, it was possible to measure a reduction in the number of GABA/central benzodiazepine receptors in the hippocampus of patients with mesial temporal lobe epilepsy caused by unilateral hippocampal sclerosis. [11C]Flumazenil has also been used to estimate the synaptic density of benzodiazepine in persons who became blind. Compared with sighted controls, a significantly lower benzodiazepine density was reported in the cerebellum of the blind subjects.
1.5 Dopaminergic System
The dopaminergic system plays a major role in neurological and psychiatric disorders such as PD, Huntington’s disease, tardive dyskinesia, and schizophrenia. The neurotransmitter dopamine plays an important role in the mediation of movement, cognition, and emotion. Changed levels of dopamine also play a role in various neuropsychiatric disorders, such as autism, attention deficit hyperactivity disorder, and drug abuse. Knowledge on changed dopamine synthesis, receptor densities, and status is important for understanding the mechanisms underlying the pathogenesis and therapy of these diseases (Elsinga et al. 2006).
To investigate presynaptic function, PET and SPECT tracers have been developed to measure dopamine synthesis and transport. For measuring dopamine synthesis, the most commonly used tracer is 6-[18F]FDOPA, whereas for dopamine transport, several radiolabeled tropane analogs are used in the clinic. Postsynaptically, dopamine exerts actions through several subtypes of the dopamine receptor. The dopamine receptor family consists of five subtypes D1–D5. In order to investigate the role of each receptor subtype, selective and high-affinity PET radioligands are required. To a lesser extent, work has been published related to radioiodinated tracers for SPECT. For the dopamine D1 subtype, the most commonly used ligand is [11C]SCH23390 or [11C]NNC112 (Kosaka et al. 2010), whereas for the D2/D3 subtype, [11C]raclopride is a common tracer. [18F]Fallypride is a suitable PET tracer for the investigation of extrapyramidal D2 receptors. For SPECT, [123I]IBZM is commercially available. For the other subtypes, no suitable radioligands have been developed yet (Tissingh et al. 1997).
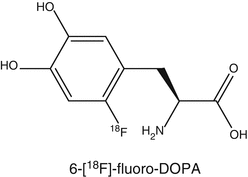
6-[18F]Fluoro-L-DOPA (FDOPA) is used to evaluate the central dopaminergic function of presynaptic neurons (Eidelberg et al. 1990; Sioka et al. 2010). Uptake of FDOPA is an indicator of DOPA transport into the neurons, its decarboxylation by amino acid decarboxylase (AADC) to 6-[18F]fluorodopamine, and capacity of dopamine storage mainly in striatum. 6-[18F]Fluorodopamine can be converted by catechol-O-methyltransferase (COMT) to 3-O-methyl-6-[18F]fluoro-L-DOPA, which is uniformly distributed throughout the brain. 6-[18F]Fluorodopamine is also metabolized via monoamine oxidase to yield 6-[18F]fluoro-3,4-dihydroxyphenylacetic acid and subsequently by COMT to yield 6-[18F]fluorochomovanillic acid. In clinical studies, AADC is commonly inhibited with carbidopa. As a result of this inhibition of peripheral AADC, there is increased availability of FDOPA in the brain.
The first FDOPA PET study of human brain was reported in 1983 showing increased accumulation of radioactivity in the striatum. In patients with established bilateral PD, FDOPA PET showed influx constant reductions in the caudate, putamen, striatal nigra, and midbrain. The decline in FDOPA uptake was more rapid in PD than normal subjects. FDOPA PET is a good tool to monitor progression of PD and therapies.
1.5.1 Dopamine Transporter (DAT)
DAT is another important target for investigation of presynaptic dopaminergic function (Brooks 2010). The function of DAT is critical for the effects of antidepressant drugs. The use of DAT ligands has a practical benefit. In 6-[18F]FDOPA PET, for example, medication with L-DOPA or other drugs is usually stopped before the PET scan, and patients sometimes receive inhibitors for AADC and COMT to reduce the peripheral metabolism of the tracer. Usually, these pretreatments are not needed for PET scans with DAT tracers. Of course, effects of other medication on DAT images should take into account.
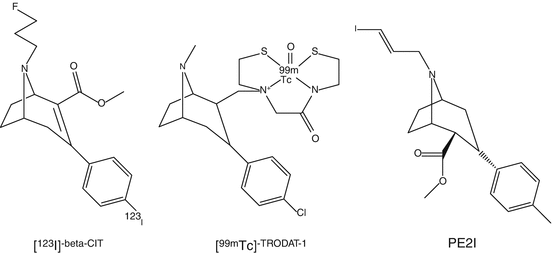
Several PET ligands of different chemical classes have been investigated to study DAT. The first reported tracers were [11C]nomifensine and [18F]GBR13119 followed by [11C]d–threo-methylphenidate. Later, tropane analogs have been developed. [11C]-β-CFT and [11C]-β-CIT showed high affinity and were metabolically stable. The major drawback of [11C]-β-CIT was its low selectivity toward DAT, although [123I]-β-CIT is clinically being used as a SPECT tracer known as DaTSCAN (Nocker et al. 2012) and was developed in the early 1990s (Innis et al. 1991). [123I]-β-CIT is commercially available. A significant correlation of severity and duration of PD with uptake of [123I]-β-CIT in striatum has been reported. [123I]-β-CIT scan is a useful tool in daily clinical practice to confirm diagnosis of PD and to differentiate from other diseases. [123I]-β-CIT SPECT can contribute to treatment selection and can be used to monitor the effectiveness of the therapies of patients. In later years, an [18F]analog of β-CIT has been developed. [18F]FP-CIT PET images of striatal uptake were superior than those of [123I]-β-CIT obtained with SPECT. Plasma analysis using [18F]FP-CIT indicated the presence of only one minor metabolite. [18F]FP-CIT can become a useful tracer in clinical practice as is [123I]-β-CIT (Sihver et al. 2007).
In order to circumvent the disadvantages, i.e., limited availability of cyclotron-produced radionuclides, 99mTc-labeled DAT tracers were developed (Kung et al. 1996). These tracers contain neutral lipophilic complexes that contain N-(alkylthiolate)tropane, aminobis(ethylthiolate), and a complex of 99mTc. [99mTc]TRODAT-1 is a tracer that belongs to this group (Chen et al. 2013). It was demonstrated that using [99mTc]TRODAT-1 scintigraphy, the loss of DAT in PD patients could be measured very accurately. [99mTc]TRODAT-1 has been successfully used to correlate striatal DAT with therapy of patients with attention deficit hyperactivity disorder (ADHD). Patients with ADHD with elevated striatal DAT responded better to methylphenidate therapy than those with low DAT levels. On the basis of these results, the possible use of DAT measurements to predict a response to methylphenidate therapy was suggested. As a follow-up result of SPECT with [99mTc]TRODAT-1, decrease of DAT levels in ADHD patients who underwent methylphenidate therapy correlated with an improvement in clinical symptoms. Caution has to be made in the interpretation of [99mTc]TRODAT-1 scans since its uptake is affected by age and sex (Mozley et al. 2001).
Other tropane tracers have increasingly been used as a biomarker for the integrity of presynaptic dopaminergic nerve cells in patients with movement disorders. 123I-labeled N-(3-iodoprop-2E-enyl)-2-β-carbomethoxy-3β-(4-methylphenyl) nortropane, named PE2I, has about ten-fold higher in vitro selectivity for the DAT than for the serotonin transporter (SERT) compared to DaTSCAN (Ziebell 2011). Furthermore, [123I]PE2I has faster kinetics than [123I]-β-CIT. Because of its fast kinetic properties, quantification of [123I]PE2I binding to DAT is possible using kinetic or graphical analysis. Since [123I]PE2I is a selective SPECT DAT radioligand with optimal kinetic properties for accurate quantification of the DAT availability in striatum, it is currently considered the best radioligand for imaging the DAT in the human brain with SPECT. More recently, 11C-methyl and the 18F-fluoroethyl analogs have been developed for human use. Studies in nonhuman primates concluded that kinetics and metabolic behavior of [18F]FE-PE2I were more favorable than for [11C]PE2I. From all studies, it was shown that uptake in striatum was highest. [11C]PE2I has been used in several human PET studies comparing controls with myoclonic epilepsy or addicted counterparts. Application of various kinetic modeling procedures showed higher DAT activity in controls. The radioactive half-life of 11C might be too short in comparison with [18F]FE-PE2I for proper estimation of striatal distribution volume (Seki et al. 2010).
1.5.2 D1 Receptor
Two PET ligands, [11C]NNC112 and [11C]SCH23390, have been developed for human application (Catafau et al. 2010; Farde et al. 1987). In a PET study of D1 receptor distribution in human brain, [11C]NNC112 showed major localization of radioactivity in the striatum and neocortex. The striatum/cerebellum and neocortex/cerebellum ratios were 3.8 and 1.8, respectively. In a study in patients with schizophrenia in comparison with normal subjects, it was shown that the binding potential of [11C]NNC112 was significantly elevated in the dorsolateral prefrontal cortex of patients with schizophrenia compared with the controls (Abi-Dargham et al. 2002).
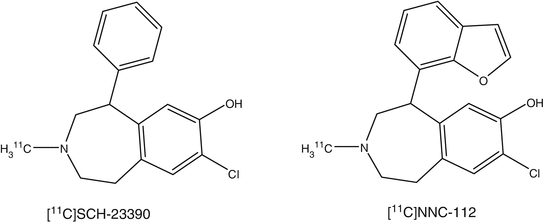
PET studies of D1 receptor distribution in human brain were reported with [11C]SCH23390, displaying most radioactivity accumulation in striatum. Striatum to cerebellum ratio and kinetic constants are commonly used as parameters. PET scans of schizophrenic patients were similar to those obtained in the normal controls (Karlsson et al. 2002). With [11C]SCH23390, it was possible to assess dopamine receptor occupancies in the striatum of patients treated with antipsychotics. Binding potential of the D1 receptors in the striatum and frontal cortex decreased with age. There was no gender difference in D1 binding potentials. In a study in patients with Huntington’s disease, a significant reduction in both the D1 ([11C]SCH23390) and D2 ([11C]raclopride) binding potentials in the striatum was shown. A comparative study using [11C]NNC112 and [11C]SCH23390 was performed in patients with schizophrenia and age-matched controls. The D1 binding potential of both tracers in the frontal cortex, anterior cingulate, temporal cortex, and striatum of the schizophrenia patients was significantly lower than those of controls (Kosaka et al. 2010).
1.5.3 D2 Receptor
Mapping of the D2 receptor in the striatum has been one of the major targets of PET research. [11C]N-methylspiperone, an analog of butyrophenone neuroleptics, was one of the first radioligands. Compared to [11C]N-methylspiperone which also has affinity for the 5-HT2 receptor, [11C]raclopride has higher selectivity for D2 receptors. Nowadays, [11C]raclopride has been used as the standard D2 receptor ligand, which binds reversibly to D2 receptors, which is an ideal property for estimation of B max (Sioka et al. 2010; Elsinga et al. 2006). Several lines of evidence indicated that [11C]raclopride binding is reduced when the synaptic concentration of endogenous dopamine is increased . For the measurement of extrastriatal D2 receptors that are expressed in much lower densities than in the striatum, ligands are required with very high affinity to achieve sufficient binding to D2 receptor. For this purpose, [11C]FLB457, which has extremely high affinity toward the receptor, was developed (Narendran et al. 2011a). Another high-affinity benzamide, [18F]fallypride, was also developed for this purpose. The longer half-life of this 18F-labeled ligand enabled quantification of D2 receptors in the caudate and putamen using a prolonged scan protocol up to 2 or 3 h (Millet et al. 2012). It is noticed that the compounds within the benzamide class, such as raclopride, FLB457, and epidepride, do not only bind to D2 receptors but also bind to D3 receptors.
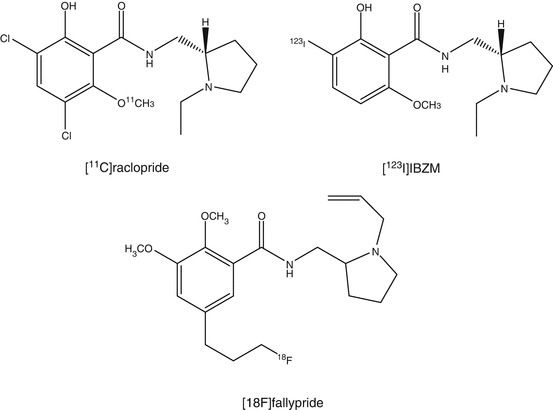
[123I]IBZM (Kung et al. 1990) is a commercially available SPECT tracer for D2/D3 receptors. Comparative studies with [11C]raclopride mention a good correlation in regional cerebral distribution. Absolute numbers such as determination of receptor occupancies measured by both tracers result in systematic differences, which can be attributed to either the analysis method or the imaging modality (Catafau et al. 2009).
1.5.4 D2/D3 Agonists
The use of agonists as imaging tracers may offer several advantages because they only bind to the high-affinity state of the receptor, whereas the antagonist tracers bind to both the high- and low-affinity binding sites. (–)-[11C]-N-propylnorapomorphine (Narendran et al. 2011b) and (+)-[11C]PHNO (4-propyl-9-hydroxynaphthoxazine) (Willeit et al. 2006) can be used to measure the functional high-affinity state of the D2 receptor. After exerting effects on second messengers, agonists will dissociate from the receptor because of a conformational change back to the low-affinity state. In comparison to antagonists, receptor binding of radiolabeled agonists is expected to be more sensitive to changes in endogenous dopamine levels that compete in binding to the receptor. As has been discussed in the section on dopamine synthesis and transport, the dopamine level is an important parameter in neurological and psychiatric diseases.
It was shown that binding of (–)[11C]-N-propylnorapomorphine was more sensitive to alterations in endogenous dopamine levels than [11C]raclopride.
A suitable D2/3 agonist PET tracer is (+)-[11C]PHNO. Its structure is based on naphtoxazine. (+)-[11C]PHNO has been evaluated in several species and in humans. PHNO displayed higher binding potential values compared to [11C]-N-propylnorapomorphine. The affinity of (+)-[11C]PHNO is higher for D3 receptors than for D2 receptors. (+)-[11C]PHNO is already used in studies of neurological diseases (Graff-Guerrero et al. 2008).
1.6 Beta-Amyloid Deposition
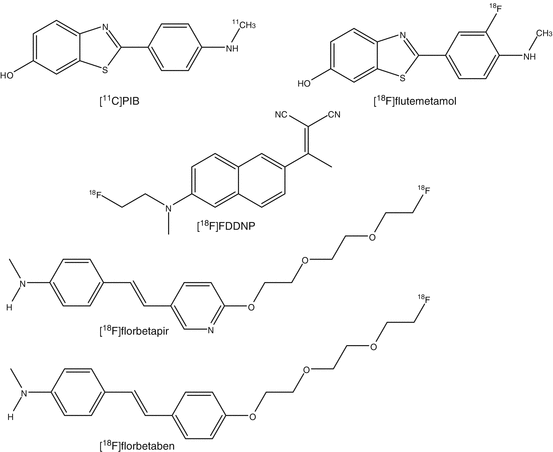
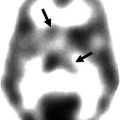
In AD, many advances have been made to understand the involved neuropathological processes. The accumulation of beta-amyloid is a primary pathological event leading to formation of neurofibrillary tangles and loss of synapses and neurons (Huang and Mucke 2012). The first clinically specific tracer for beta-amyloid imaging was 11C-labeled Pittsburgh compound B (PIB) (Klunk et al. 2004). PIB is an analog of thioflavin T that binds to fibrillar beta-amyloid deposits with high sensitivity and specificity. PIB binds to both extracellular amyloid plaques and vascular amyloid deposits. At tracer dosages, PIB does not bind to neurofibrillary tangles or Lewy bodies (Zhang et al. 2012).
To improve the accessibility of beta-amyloid imaging with PET, a second generation of 18F-amyloid tracers was developed. Four 18F-amyloid imaging agents are in advanced stages of development: flutemetamol, a 3′-fluoro analog of PIB (Thurfjell et al. 2012); florbetapir, a styrylpyridine derivative (Doraiswamy et al. 2012); FDDNP, a naphthol analog (Ossenkoppele et al. 2012); and florbetaben, a derivative of stilbene (Barthel and Sabri 2011). With the exception of FDDNP, these tracers show comparable results to PIB in clinical populations, although nonspecific white matter binding appears to be higher.
Besides these three [18F tracers, a few more ligands have been evaluated in man. A human study with [11C]BF-227 showed that retention of radioactivity is higher in brain regions that are known to contain beta-amyloid plaques in AD patients than in control subjects (Furukawa et al. 2010). No significant difference was observed in any brain regions between young and aged normal subjects. 2-(1-(6-[(2-[18F]Fluoroethyl)(methyl)amino]-2-naphthyl)ethylidene)malononitrile ([18F]FDDNP) showed higher binding in AD brain than those of healthy controls. Despite of its slow clearance kinetics for PET imaging, [18F]FDDNP is used for detection of both neurofibrillary tangles and beta-amyloid plaques in patients with AD. In a comparative study between [11C]PIB and [18F]FDDNP, cortical binding potential of [11C]PIB showed higher binding in patients with AD than in controls and patients with mild cognitive impairment (MCI). [18F]FDDNP uptake was higher in brains of AD patients than in healthy controls, but MCI could not be distinguished from AD or from controls. Differences in binding potentials between patients with AD, or MCI, and healthy controls were more pronounced for PIB.
1.7 NMDA Receptor, Glycine Transport
Glycine acts as a neurotransmitter and is a modulator of the neuroexcitatory activity of the N-methyl-d-aspartate (NMDA) receptor. Impaired function of the NMDA receptor is responsible for cognitive dysfunction in patients suffering from neuropsychiatric diseases, like schizophrenia and related disorders (Zorumski and Izumi 2012). Specific transporters are responsible for the uptake of glycine into the brain. The high-affinity transporters, GlyT-1 and GlyT-2, block the activity of glycine on the NMDA receptor in the synapse. GlyT-1 has been shown to maintain low levels of glycine at the synapse. Inhibition of GlyT-1 would increase glycine concentrations around the synapse, resulting in enhanced activity of the NMDA receptor. Several imaging agents for GlyT-1, such as [18F]-2,4-dichloro-N-((1-(propylsulphonyl)-4-(6-fluoropyridine-2-yl)piperidine-4-yl)methyl)benzamide ([18F]MK-6577 and [11C]GSK931145 were developed. Although the tracer is slowly metabolized, [11C]GSK931145 has been successfully evaluated for the in vivo visualization of GlyT-1a humans (Gunn et al. 2011).
1.8 P-Glycoprotein
Permeability of the blood–brain barrier (BBB) is one of the factors of protection of the brain against harmful compounds (Bartels 2011). The BBB only allows entry of lipophilic compounds with low molecular weights by passive diffusion. In addition, there are transporters such as P-glycoprotein (P-gp), multidrug resistance-associated protein (MRP), and organic anion-transporting polypeptides (OATPs). The action of these carrier systems results in rapid efflux of harmful compounds from the central nervous system (CNS). P-gp is the most studied transporter with PET and SPECT. PET studies related to P-gp were directed to (i) direct evaluation of the effect of P-gp modulators on the cerebral uptake of therapeutic drugs, (ii) assessment of mechanisms underlying drug resistance in epilepsy, (iii) examination of the role of the BBB in the pathophysiology of neurodegenerative and affective disorders, and (iv) exploration of the relationship between polymorphisms of transporter genes and the pharmacokinetics of test compounds within the CNS (Colabufo et al. 2010; Elsinga et al. 2005).
Several radiotracers have been prepared to study P-glycoprotein function in vivo with PET or these tracers unintendedly proved to be P-gp substrates. These include alkaloids ([11C]colchicine), anticancer drugs ([11C]daunorubicin, [18F]paclitaxel, [11C]tariquidar, [11C]elacridar), calcium antagonists ([11C]-R-(+)-verapamil), ß-adrenoceptor antagonists ([11C]-(S)-carazolol, [18F]-(S)-1′-fluorocarazolol, [11C]carvedilol), serotonin 5-HT1A receptor antagonists ([18F]MPPF), opioid receptor antagonists ([11C]loperamide, [11C]carfentanyl), and various 64Cu-labeled copper complexes. By far, [11C]verapamil is the most investigated PET tracer for P-gp. The tracer has been administered to healthy volunteers and blocking studies with the immunosuppressant agent cyclosporin A were carried out. The results indicated that P-gp activity at the human BBB can be measured despite its high lipophilicity. Using the distribution volume as parameter, [11C]verapamil uptake in the midbrain was significantly increased (18 %) in Parkinson’s disease patients compared with healthy controls. This suggests a decrease in P-gp activity at the BBB of PD patients (Bartels et al. 2010).
Loperamide is an opiate agonist and a substrate for P-gp at the BBB. [11C]Loperamide ([11C]Lop) has been applied for studying P-gp function and multidrug resistance in tumors and normal tissues noninvasively. However, demethylation of [11C]Lop to [N–methyl–11C]-N-desmethyl-loperamide ([11C]dLop) hampers its use as PET tracer for P-gp function since [11C]dLop is also a good substrate for P-gp. Therefore, [11C]dLop has been studied as a PET tracer for studying P-gp function (Seneca et al. 2009). In a study with healthy volunteers, there was minimal brain uptake of [11C]dLop. There were five hydrophilic radiometabolites. Because of much lower nonspecific binding, [11C]dLop is preferred over [11C]verapamil.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


