div class=”ChapterContextInformation”>
4. Atherosclerosis of the Carotid Artery
Keywords
CarotidAtherosclerosisLipidsImmune cellsVulnerable plaquesRisk factorsPathogenesis of Atherosclerosis
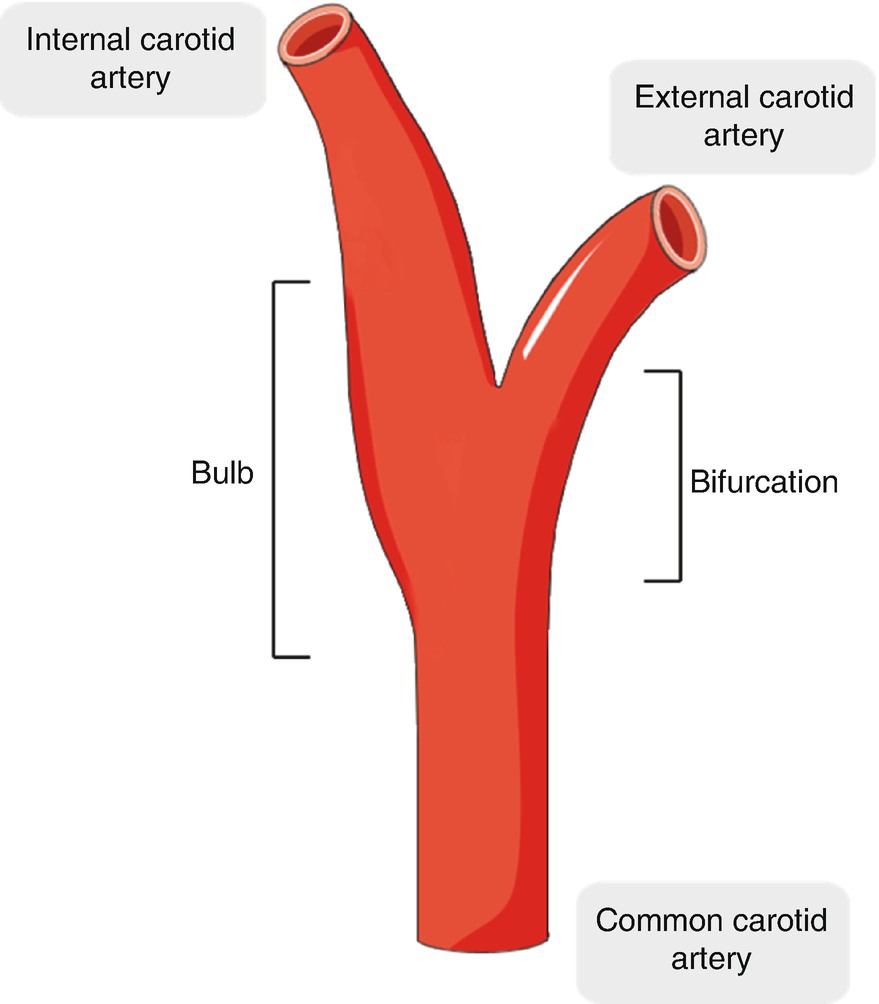
Atherosclerosis is a two-faced disease. While it has one of the longest incubation periods known to man, its main complications, myocardial infarction and ischemic stroke, happen suddenly and often without prior warning. The earliest signs of disease, fatty streak lesions, can already be found in the first decade of life in the aorta and will develop in the coronary arteries in the second decade [1]. These lesions form after the arterial endothelium is activated by an injurious stimulus, such as low shear stress, turbulent flow, or oxidative stress. Both low shear stress and turbulent flow are present at arterial bifurcations, including the bifurcation of the internal and external carotid artery, causing a high incidence of atherosclerotic plaques at sites like these [2].
Due to the activation of the endothelium, lipid components, most notably low-density lipoproteins (LDL), start accumulating in the subendothelial space. Once trapped in the subendothelial space, the lipid particles are altered, most importantly by lipid oxidation as a result of exposure to the oxidative waste of vascular cells. The oxidation of LDL particles leads to the formation of oxidized LDL (oxLDL), the most potent early stimulator of the inflammatory process driving atherosclerosis [3]. The activated endothelium also starts attracting immune cells by expressing cell adhesion molecules and secreting chemokines, a process that is also propagated by immune cells, mostly macrophages, already present in the plaque [4, 5].
The extent and speed of atherosclerotic plaque buildup are modulated by the severity of dyslipidemia as well as severity of the inflammatory response, and both are correlated with incidence of cardiovascular disease [6]. Many factors play a role in the severity of the inflammatory response in atherosclerosis; some of the most notable are mechanical forces [2], sex hormones [7], chronic inflammatory diseases [8], metabolic syndrome/obesity [9], (epi)genetics [10, 11], and diabetes mellitus [12].
Lipids
More than a century ago, the German pathologist Rudolf Virchow discovered that cholesterol was present in atherosclerotic plaques. In the 1950s, the Framingham Heart Study firmly established high lipid levels as a risk factor for atherosclerosis [13]. Lipid-lowering statins (inhibitors of HMG-CoA reductase) are very successful in reducing cardiovascular risk [14]. PCSK9 inhibitors are a new class of drugs, capable of dramatically lowering lipids in combination with statins, resulting in the further lowering of cardiovascular risk [15, 16]. The importance of lipids is also evident in mouse models, where, besides a knockout of genes such as apolipoprotein E (ApoE) or the LDL receptor (LDLR), a lipid-rich diet is needed to induce significant atherosclerosis.
Intracellular cholesterol homeostasis is the cumulative result of three simultaneous processes: (1) intracellular synthesis; (2) uptake from plasma lipoproteins, most importantly via LDL; and (3) reverse cholesterol transport by high-density lipoproteins (HDL). Cholesterol levels are sensed by the LXR (liver X receptor), a nuclear receptor which stimulates cholesterol transport toward the liver, biliary excretion, and possibly reduction in cholesterol synthesis. Contrary to the function of LXR, sterol regulatory element-binding protein 1c and 2 (SREBP-1c and SREBP-2) increase lipogenesis, cholesterol synthesis, and cholesterol uptake. Mice lacking the LXR gene feature a higher expression of SREBP-2, as well as several cholesterol-related genes such as HMG-CoA synthase and HMG-CoA reductase [17]. These properties make increasing LXR or decreasing SREBP interesting new therapeutic targets.
Low-Density Lipoproteins
Low-density lipoproteins are a large contributor to cardiovascular risk, and lowering LDL is one of the most effective ways for risk reduction [14]. In patients, both in clinics and clinical trials, plasma levels of LDL are generally not measured directly due to high costs; instead, LDL-C is determined using the Friedewald equation (LDL-C = Total cholesterol – HDL – triglycerides). In most situations, LDL and LDL-C correlate well; however, in certain situations, most notably hypertriglyceridemia, the LDL-C value can deviate from the actual LDL concentration [18]. In health, cells upregulate the LDL receptor (LDLR) when in need of cholesterol, which is required for cell growth, steroid/hormone synthesis, and the structural integrity of cell membranes. Circulating LDL particles bind to the LDL receptor and are then transported into the cell via endocytosis. At higher plasma levels of LDL, the LDLR will become saturated. When the amount of circulating LDL exceeds the ability of the liver to catabolize LDL, receptor-independent uptake of LDL will occur, especially at sites where the endothelium has become activated, ultimately leading to the formation of foam cells and fatty streaks [18]. After binding to LDL and internalizing it, the LDLR is able to be reused on the cell membrane. The enzyme PSCK9 inactivates the LDLR, making it unable to be reused. Inhibiting the PSCK9 enzyme severely lowers plasma LDL-C levels [15, 16].
Oxidized LDL
When entering the vessel wall, LDL is modified via a variety of mechanisms. The most relevant mechanism is the oxidation of LDL by reactive oxygen species (ROS). While non-modified LDL binds to the LDLR, oxLDL binds to scavenger receptors including CD36, SR(A), and LOX-1 that are mostly expressed by macrophages [3]. OxLDL is a potent activator of macrophages, ultimately leading to the formation of foam cells. The presence of oxLDL upregulates the LOX-1 receptor while downregulating the LDLR, initiating a positive feedback loop where more LDL is oxidized and then taken up via LOX-1 [18].
Triglycerides
Like LDL, triglycerides have been shown to be an important risk factor for cardiovascular disease in the Framingham Heart Study [13]. In humans, triglycerides are transported via very low-density lipoproteins (VLDL) [18]. Pharmacological inhibition of triglycerides has not been as successful as LDL lowering. However, some recent studies have shown that in patients on statins with high triglycerides, triglyceride lowering reduces major adverse cardiovascular events (MACE) [19].
HDL
HDL is the smallest of the lipoprotein family. HDL acts as a reverse carrier of cholesterol, as it is able to uptake cholesterol from cells. HDL is important for the recycling of cholesterol back to the liver and the delivery of cholesterol toward hormone-producing organs [19]. As higher levels of HDL are correlated with lower incidence of adverse cardiovascular events in the general population, HDL was thought of as a promising new therapeutic target [13]. However, this has come under scrutiny in recent years as more in-depth examinations of population data show no other associations between HDL and cardiovascular events, except for an increased risk when HDL levels are very low. Furthermore, in Mendelian randomization studies, it was shown that genetically high HDL does not protect against cardiovascular events [20]. Ultimately, increasing HDL through various pharmacological methods in cardiovascular patients does not result in the lowering of cardiovascular risk [20, 21].
Inflammation
Monocytes
Among the first cells recruited to the activated endothelium are monocytes. Monocytes are short-lived mononuclear phagocytes, which differentiate into macrophages once they infiltrate into the vessel wall. Monocytes are recruited to the site of the plaque by the secretion of chemokines such as CCL2 and CCL5 by the endothelium. The endothelial cells express the adhesion molecules P-, L-, and E-selectin, ICAM-1, and VCAM-1, to which the monocyte adheres. After adhering to the vessel wall, the monocytes start rolling along the activated endothelium and subsequently enter the subendothelial space, either via para-endothelial or trans-endothelial migration [4, 5].
In mice, two general subpopulations of monocytes have been described: the inflammatory Ly6Chigh monocyte, which is preferentially recruited toward inflamed tissue, and the patrolling Ly6Clow monocytes which are homed toward non-inflamed tissues. In murine atherosclerosis, inflammatory (Ly6Chigh) monocytes are preferentially recruited toward the vessel wall and have been shown to aggravate atherosclerosis [22–24]. In humans, three major monocyte subsets have been described. Similar to mice, human monocytes can be divided into inflammatory CD14++CD16- expressing monocytes (classical monocytes) and anti-inflammatory CD14+CD16++ expressing monocytes (nonclassical monocytes). Additionally, in humans, an intermediate CD14++CD16+CCR2+-expressing subset (intermediate monocytes) can be identified. Functionally, CD14++CD16- monocytes (classicals) support inflammation and phagocytosis, CD14++CD16+CCR2+ monocytes (intermediates) are pro-inflammatory, and CD14+CD16++ monocytes (nonclassicals) are aimed toward restoration of homeostasis (healing) [5, 25]. In humans, both higher levels of classical monocytes and intermediate monocytes have been shown to correlate with a higher incidence of cardiovascular disease [24, 26, 27].
When monocytes migrate over the endothelial barrier toward the intima, they come into contact with colony-stimulating factors, most importantly macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by endothelial cells, vascular smooth muscle cells (vSMC), and macrophages. These factors initiate the differentiation of monocytes into macrophages [28].
Macrophages
In healthy tissue, the most notable macrophage functions are phagocytosis (the uptake of solid particles or pathogens), efferocytosis (the uptake and clearance of apoptotic cells), tissue remodeling, and chemoattraction. All of these processes are impaired in the atherosclerotic plaque.
Like monocytes, macrophages can be divided into different subtypes. Which subtype a macrophage becomes is mostly determined by the environment and stimuli the macrophage is exposed to. At the extremes, macrophages can be divided into the pro-inflammatory M1- and the anti-inflammatory M2 macrophage. M1 macrophages are induced by Th1 cytokines and excrete pro-inflammatory cytokines like TNF, IL-6, IL-1β, and IL-12. Sustained M1 activation results in tissue damage and eventually impaired wound healing. The anti-inflammatory M2 macrophages are induced by Th2 cytokines and excrete anti-inflammatory cytokines like IL-10 and TGF-β. Functionally M2 macrophages scavenge debris and promote the resolution of inflammation, most importantly by clearing apoptotic cells (efferocytosis) and the dampening of immune responses, ultimately promoting tissue repair and healing [29].
It is important to note however that the M1 vs. M2 division is a simplified concept. Between the M1 and M2 phenotypes, a spectrum of other macrophage phenotypes can be identified, each with a different or yet undetermined function. In experimental mouse models of atherosclerosis, the M1 subtype and the plaque-specific M4 subtype are pro-inflammatory and pro-atherogenic. An increase of these macrophage subtypes inside the murine atherosclerotic plaque is correlated with larger and more vulnerable plaques. The other macrophage subtypes, the M2 phenotype and the plaque-specific M(hb) and Mhem phenotypes, have been shown to be anti-inflammatory and anti-atherogenic in murine atherosclerosis. The Mox phenotype that has been identified fairly recently is currently not well understood [30]. Through phagocytosis, macrophages take up oxLDL, an important step in the progression of atherosclerosis. Via scavenging receptors CD36, SR (A), and LOX-1, oxLDL is transported into the macrophage, where it is transformed into cholesteryl esters by the enzyme acetyl-CoA acetyltransferase (ACAT1).
Foam Cells
The excessive accumulation of cholesteryl esters in the endoplasmic reticulum of the macrophage induces its transformation to a foam cell (named after its foamy appearance on histology) [3]. Furthermore, the phagocytosis of cholesterol crystals activates the NLRP3 inflammasome, which in turn induces an enhanced secretion of IL-1β. The inhibition of IL-1β via the monoclonal antibody canakinumab has been shown to reduce cardiovascular events in patients [31, 32].
It is currently unknown which specific macrophage phenotype is the precursor of the foam cell. However, several studies have shown that the M2 phenotypes are more prone to differentiate into a foam cell.
The transformation of a macrophage into a foam cell induces various apoptotic pathways that ultimately lead to its demise. In advanced atherosclerosis, efferocytosis is impaired, leading to the formation of “post-apoptotic” foam cells that ultimately undergo secondary necrosis. This process is one of the main drivers of the formation of the necrotic core and a major cause of enhanced and sustained inflammation in the plaque [33].
Neutrophils
Neutrophils are short-lived phagocytic cells. Even though few neutrophils can be found in advanced atherosclerotic plaques, their importance in atherosclerosis is not to be underestimated. The degree of hypercholesterolemia-induced neutrophilia is positively correlated with atherosclerotic plaque size in experimental models and associated with plaque vulnerability in humans [34]. In both health and atherosclerosis, neutrophils work in conjunction with macrophages. Macrophages improve neutrophil survival through secretion of growth factors such as G-CSF, GM-CSF, and TNF. In turn, neutrophils attract monocytes toward the plaque and improve endothelial permeability, ultimately aiding and increasing monocyte infiltration into the atherosclerotic plaque [35]. Neutrophils, co-localized with macrophages, can be found at the cap of the atherosclerotic plaque. There, neutrophils release granule proteins, matrix metalloproteinases, and cathepsins. These factors are potent determinants of atherogenesis in experimental animal models, and many of them have been correlated with vulnerable plaques [36, 37].
T Cells
T cells play a central role in cell-mediated immunity. These immune cells are defined by T cell receptor (TCR) on their cell surface. Antigen-presenting cells (APCs, DCs, macrophages, and B cells) present antigens via MHC II to the TCR, causing the T cell to become activated after simultaneous interaction of co-stimulatory molecules with their ligands, though it is also possible for T cell activation to be antigen independent.
Several subtypes of T cells have been identified, the T-helper cell (Th), the cytotoxic T cell (Tc), and the regulatory T cell (Treg). Like macrophages, T cells can somewhat switch between these subtypes depending on environmental cues it encounters.
T-helper cells (Th cells) are defined by the expression of CD4 and functions as an assistant to other immune cells. Most importantly, it induces activation of B cells, macrophages, and cytotoxic T cells and maturation of B cells. In general, there are two subtypes of Th-cells, Th1 and Th2. The Th1 subtype is considered pro-inflammatory; the Th2 is more anti-inflammatory. The balance between Th1 and Th2 levels plays a role in several diseases, like psoriasis (Th cell population skewed toward Th1) and asthma (Th-cell population skewed toward Th2) [38, 39].
T cells are recruited toward the vessel wall via mechanisms similar to macrophage recruitment and make up about 10% of the total cell populace in an atherosclerotic plaque. About 70% of these T cells are T-helper (CD4+) cells, the remaining fraction mostly being cytotoxic (CD8+) T cells [40]. In the vessel wall, T cells become activated in response to antigens and start to produce inflammatory agents (e.g., IFN-γ).The antigen that triggers the immune response and induces T cell proliferation and polarization is most likely atherosclerosis-specific antigens such as (the ApoB100 part of) LDL [41].
The majority of CD4+ T cells, and thus the majority of T cells, are of the Th1 subset. This subset has been extensively described as pro-atherogenic. Through the production of IFN-γ, T cells recruit macrophages and other T cells toward the atherosclerotic plaque, causing an increase in the secretion of Th1-promoting cytokines, an increased lipid uptake by macrophages, and an increase in foam cell formation, ultimately leading to an increased plaque growth and vulnerability. The positive feedback loop where Th1 cells recruit other Th1 cells is a major contributor to plaque progression. In patients suffering from cardiovascular disease, higher levels of Th1 cells have been found [42–44].
Th2 cells support antibody production of B cells via secretion of IL-4, IL-5, IL-10, and IL-13. While the Th2 subtype is increased in hyperlipidemia, the Th2 subtype is rare in atherosclerotic lesions. The role of Th2 cells in atherosclerosis is still poorly understood and seemingly contradictory. While IL-5 and IL-13 clearly protect against atherosclerosis, the prototypic Th2 cytokine IL-4 doesn’t protect against atherosclerosis [42–44].
Regulatory T cells comprise around 5% of CD4+ T cells in the peripheral blood in humans. An overall protective role of regulatory T cells is suggested in both murine and human atherosclerosis studies. In mice, depletion of regulatory T cells aggravated atherosclerosis, suggesting that regulatory T cells limit plaque inflammation and thus disease progression. The mechanism of this action has not yet been identified. In humans, relatively low levels of regulatory T cells were found in atherosclerotic plaques, which might play a role in the persistent inflammation in these lesions. Moreover, compared to stable plaques, in rupture-prone plaques, fewer regulatory T cells can be found. Together, this data suggests a protective role of regulatory T cells in atherosclerosis [45].
In early atherosclerosis, few cytotoxic (CD8+) T cells are present in the atherosclerotic plaque. However, they appear to be the dominating immune cell in advanced human plaques. In these lesions, CD8+ T cells concentrate around shoulder regions and fibrous caps. Furthermore, in murine atherosclerosis, CD8+ T cells have a pro-atherogenic effect. CD8+ T lymphocytes most likely promote the development of vulnerable atherosclerotic plaques by inducing apoptosis of macrophages, smooth muscle cells, and endothelial cells that in turn leads to secondary necrosis and necrotic core formation [46].
B Cells
B cells are known for their ability to produce antigens and are identified by the presence of a B cell receptor on their cell membrane. Besides antigen production, B cells are also able to serve as antigen-presenting cells and can activate both CD4+ and CD8+ T cells. Furthermore, they can promote chemokine production, produce a variety of cytokines (e.g., IFN-γ, IL-2, IL-12, IL-4, IL-6, and IL-10), promote leukocyte recruitment, and polarize T cells. Mature B cells can be divided into two categories: the B1 subtype, which is innate, T cell independent and produces polyreactive natural IgM antibodies, and the B2 subtype, which is important in adaptive immunity, as it is able to produce specific IgG antibodies tailored to a specific antigen and T cell dependent. Natural IgM antibodies against oxLDL, produced by B1 cells, are protective against atherosclerosis. In murine atherosclerosis, IL-5 mediates the production of the anti-oxLDL IgM antibodies. In contrast to the IgM antibodies produced by B1 cells, IgG antibodies against oxLDL promote atherosclerosis. This is evident in mice lacking B2 cells, as those have decreased plaque formation. Conversely, an increase in B2 cells increased plaque formation. This correlates with the fact that increased titers of anti-oxLDL antibodies are associated with an increase in adverse cardiovascular events [47].
Progression Toward the Advanced Plaque
As defined by histology, different stages of plaque development can be distinguished [48, 49]. As noted above, atherosclerotic lesions start developing around the second decade of life. The first detectable abnormalities in the vessel wall are intimal xanthomas (or fatty streaks). These lesions show accumulation of foam cells in the vessel wall, without the presence of a necrotic core or fibrous cap. Both human and animal research suggest that most of these lesions regress [50, 51].
While intimal xanthomas are detectable in children, the majority of advanced atherosclerotic lesions seem to originate from intimal thickening [52]. Intimal thickening refers to the accumulation of smooth muscle cells (SMCs) in the intima, without the presence of lipid, macrophages, or foam cells. From normal intima thickening, pathological intimal thickening can develop. The lesions are characterized by a proteoglycan-rich matrix containing smooth muscle cells, perhaps a few macrophages and other immune cells, with small areas of lipid accumulation but with minimal or no evidence of a lipid core or necrosis. These lesions seem especially prone to erosion (see below).
Both intimal xanthomas and lesions with (pathological) intimal thickening can develop into a fibrous cap atheroma. These atherosclerotic plaques are characterized by a fibrous cap, overlaying a well-developed necrotic core. The fibrous cap is an overlay of connective tissue, consisting of smooth muscle cells and collagen in a proteoglycan matrix. This fibrous cap separates the blood from the highly thrombogenic necrotic core, which consists of a core of necrotic cells, surrounded by macrophages. When the necrotic core increases in size, increasingly large amount of extracellular lipids and cholesterol crystals will accumulate in it and form one or several large masses, also called the lipid core. Depending on the thickness of the fibrous cap, plaques can be quantified as thick-cap fibroatheromas or thin-cap fibroatheromas. The cut-off value distinguishing between these two plaques is different per vascular bed. In carotid plaques, plaques with a cap thinner than 165–200 μm are considered thin-capped [53, 54], while in coronary atherosclerosis, this cut-off value is 65 μm [55].
Thick-capped fibroatheromas rarely rupture but can erode (see below). Conversely, thin-cap fibroatheromas are very prone to rupture; most ruptured coronary plaques found during an autopsy study of >200 deceased patients had a cap thinner than 65 μm [48].
Ultimately, plaques can develop into stable fibrocalcific plaques. While these plaques can still cause clinical symptoms due to a reduction of lumen size, they will not cause acute ischemic syndromes. These plaques usually cause significant stenosis, are collagen-rich, and have large calcified areas. They contain few inflammatory cells, though a necrotic core may be present [48].
It is very important to note that progression of atherosclerotic disease is not a linear process. During the lifespan of an atherosclerotic plaque, its fibrous cap thickness can increase or decrease, and the plaque can regress all together or progress into a fibrocalcific plaque, making a previously dangerous (vulnerable) plaque suddenly stable or vice versa [56].
The Vulnerable Plaque
- 1.
Plaque rupture: rupture of the fibrous cap overlaying the plaque, exposing the highly thrombogenic lipid core to blood, which immediately starts clotting, ultimately resulting in a thromboembolic stenosis or even occlusion of the vessel.
- 2.
Plaque erosion: typically, the endothelial layer is absent in these plaques, resulting in the exposure of smooth muscle cells and extracellular matrix to blood, resulting inocclusion precipitated largely by the thrombus or distal embolization.
- 3.
Calcified nodule: a relatively rare occurrence where calcifications protrude into the lumen, which are correlated with ischemic stroke [57–59].
Plaque Rupture
In a histopathological study of carotid endarterectomy, 67% of stroke patients had a thrombus at the endarterectomy site, compared to 27% in asymptomatic individuals. Plaque rupture was the most common cause of thrombus formation in both cases (64% of cases in stroke patients, compared to 25% of asymptomatic patients) [53]. Similar numbers are seen in coronary atherosclerosis, where about 60% of acute myocardial infarctions are due to plaque rupture. However, it should be noted that carotid plaque composition has changed over the years, as a decrease in percentage of carotid plaques with vulnerable characteristics has been observed. The same phenomenon can be observed in coronary atherosclerosis, where a decrease in plaque rupture and concurrent increase in plaque erosion has been observed. These changes over time are possibly due to the increased statin use and decreased tobacco use in recent years [60].
Plaques vulnerable to rupture (vulnerable plaques) have several histological characteristics, first identified in postmortem studies in patients who died from myocardial infarction. These plaques have a large lipid core (>40% of the total plaque area), a large necrotic core, intra-plaque neovascularization, intra-plaque hemorrhages, inflammatory cell infiltration into the fibrous cap, and, most importantly, a thin fibrous cap [1]. These same characteristics are of importance in carotid atherosclerosis, with one notable difference [61]. In carotid vulnerable plaques, intra-plaque hemorrhage (IPH) is especially predictive of future carotid atherosclerosis progression and stroke [62, 63]. Advanced lesions often show IPH, and it is postulated that micro-hemorrhages within the plaque occur from microvessels, originating from the adventitial vasa vasorum or from neovessels arising from the luminal endothelium. Both IPH and plaque rupture are associated with an increased density in microvessels [62].
Plaque rupture occurs when the shear stress pulling on the fibrous cap exceeds the tensile strength of this fibrous cap. In the carotid artery, retrospective studies determined that caps thinner than <165 μm [53] or <200 μm [54] are prone to rupture. These plaques are termed thin-cap fibroatheromas (TCFA). In coronary atherosclerosis, the threshold for rupture prone plaques is lower (65 μm [55]), most likely due to differences in flow and vessel diameter. The coronary threshold and the importance of the thin cap in coronary plaque rupture have been confirmed by in vivo intravascular imaging in patients [64]. In carotid atherosclerosis, no prospective studies determining the usefulness and/or accuracy of these values have been performed yet.
Thinning of the fibrous cap is induced by two concurrent mechanisms. Macrophages and foam cells inside the plaque induce tissue remodeling, especially fibrous cap thinning, via the secretion of several proteolytic enzymes such as plasminogen activators, cathepsins, and matrix metalloproteinase) [65]. Furthermore, smooth muscle cells (SMC) are gradually lost in the thinning fibrous cap due to SMC apoptosis. At the site of cap rupture, SMCs are usually absent. Both of these processes are most apparent at the shoulder regions of a plaque, which is probably why this is the region with the thinnest fibrous cap [66].
In both coronary and carotid atherosclerosis, rupture of a thin fibrous cap can sometimes be induced by a trigger, such as alcohol or coffee consumption, heavy meals, cigarette smoking, anger, anxiety, work stress, industrial accidents, war and terror attacks, earthquakes, and sporting events [67]. These triggers likely cause activation of the sympathetic nervous system, resulting in an increased heart rate and higher blood pressure and possibly increased platelet reactivity. It should be noted that some studies indicate that plaque rupture caused by these triggers would have happened a few weeks later anyway [66].
Plaque Erosion
About 35% of myocardial infarctions are due to plaque erosion, a number that is seemingly rising in the last decade [60]. These numbers might be lower in carotid atherosclerosis, being the underlying mechanism in about 10% of stroke or TIA patients, though it should be noted that this study was performed with a low number of patients in an older database [68]. Plaque erosion occurs when the endothelial cell layer overlying the fibrous cap disappears, thus exposing the thrombotic extracellular matrix to the blood, ultimately leading to clot formation. As no identifying morphological features of plaque prone to erosion have been identified, characterizing these plaques has proven difficult [57].
In regions of disturbed flow, toll-like receptor 2 (TLR2) is activated on endothelial cells. Ligation of TLR2 induces the production of IL-8, promoting neutrophil adhesion and recruitment. In vitro, co-incubation of endothelial cells with TLR2 agonists and neutrophils promotes endothelial cell death. This combination of factors might also lead to the loss of endothelial cells in vivo, ultimately resulting in the exposure of the fibrous cap to the blood. This exposure activates platelets, causing them to release pro-inflammatory mediators (further aggravating the process leading to endothelial cell death) and to release blockers of fibrinolysis (increase the durability of clots) and causing them to adhere to the exposed fibrous cap, leading to the initiation of thrombus formation [69].
Calcified Nodules
About 6–7% of carotid thrombosis is thought to be caused by calcified nodules, though it should be noted that this number is derived from a low number of cases [53]. This number is slightly lower in coronary atherosclerosis [58]. First introduced by Virmani et al., calcified nodules refer to a rare type of thrombosis caused by extruding calcified nodules into the vessel lumen [48]. These calcified nodules are more often present in older individuals and tortuous arteries. In coronary atherosclerosis, calcified nodules identified by IVUS in the PROSPECT trial were unlikely to cause adverse cardiovascular events during a 3-year follow-up [57].
Atherosclerosis in the Carotid Artery
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


