div class=”ChapterContextInformation”>
11. Pathophysiology of Coronary Artery Disease
Keywords
Coronary artery diseaseAtherosclerosisRuptureErosionCalcified noduleIntroduction
Despite many and continued advances in medical therapies and diagnostic imaging modalities and greater understanding of the molecular mechanisms of atherosclerosis, coronary artery disease (CAD) remains the major cause of morbidity and mortality throughout the world. Deaths from cardiovascular disease (CVD) reached a total of 17.3 million worldwide in 2012 and are expected to rise up to 23.6 million by 2030 [1]. Atherosclerosis is a mutifactorial disease and includes well-known risk factors like hypertension, hyperlipidemia, diabetes mellitus, smoking, and family history. In addition, recently high-sensitivity C-reactive protein, homocysteine, high fibrinogen, Lp(a), and LpPLa2 have been recognized as biomarkers associated with atherosclerosis. The American Heart Association (AHA) has suggested adding health metrics like physical activity, healthy diet, and body mass index (BMI) in addition to blood pressure, total cholesterol, blood glucose, and smoking. Understanding the morphologic features of the atherosclerotic disease as well as its background can result in better comprehension of the disease. It is thought that most acute coronary syndromes (ACS) are the result of luminal thrombosis, although not all are associated with occlusive diseases. There are three main causes of coronary thrombosis such as plaque rupture, erosion, and calcified nodule [2]. Based on the autopsy studies, the majority of thrombosis occurs from plaque rupture (55–65%), followed by erosion (30–35%) and, least frequent, calcified nodule (2–7%) [2]. In a recent systematic worldwide review of published autopsy series of the causes of coronary thrombosis, plaque rupture occurred in 73% which is higher than our data and is likely due to the inclusion of cases of acute myocardial infarction and those with known coronary artery disease. Our data is different because we have only included cases of sudden coronary death without a known history of coronary artery disease, with a mean age of 50 years. Also, individuals from hospital-based autopsies are usually older (>60 years) than those dying suddenly [3]. In addition to age distribution, plaque rupture is more common in AMI patients (79%) as compared to individuals who have died suddenly (65%) and to our knowledge does not equate the hospital. Also, the incidence of erosion is lower in AMI patients (25%) than individuals dying of sudden coronary death (35%) [2, 4].
This chapter will focus on the structural characteristic of plaque progression and mode of presentation, i.e., acute coronary syndrome versus stable angina.
The Classification of Atherosclerosis
It is important to understand how atherosclerosis progresses and what lesion constitutes atherosclerosis and to determine the various stages of lesion progression. In the mid-1990s, Stary et al. under the auspices of the American Heart Association defined early and advanced lesions of atherosclerosis in two separate publications [5, 6]. The first consensus document classified the stages of early atherosclerosis into three different numerical categories: initial lesion is the first microscopic and chemical detection of lipid deposits, Type I; fatty streak (FS) is the gross visibility of yellow color streaks, Type II; and transition or intermediate lesion is the transition to a fibroatheroma, Type III. In a separate manuscript, the definition of advanced lesions of atherosclerosis was described as fibroatheroma with well-defined region of necrotic core in the intima, Type IV; fibroatheroma or atheroma with a thick layer of fibrous connective tissue is Type V; and complicated plaques with surface defects, and/or hematoma-hemorrhage, and/or thrombosis are Type VI. At this time, other causes of coronary thrombosis, i.e., erosion and calcified nodule, did not weigh in at all in this classification.
Our laboratory with the largest registry of human autopsy cases of patients dying from sudden coronary death worldwide reported that this numeric nomenclature was not enough. For example, it was not appreciated that the precursor lesion of plaque rupture was a thin-cap fibroatheroma and that erosion and calcified nodule were other causes of coronary thrombosis. The atherosclerosis process is considered too complicated to be classified into numerical groups. These problems encouraged us to modify the AHA classification. In our modified AHA classification published in 2000, AHA types I–IV lesions were replaced by descriptive terminology, i.e., adaptive intimal thickening (AIT), intimal xanthoma (FS), pathologic intimal thickening, and fibroatheroma [2], which were later divided into early and late fibroatheroma. AHA types V and VI were discarded since these classifications did not show other types of coronary thrombosis such as erosion and calcified nodule and stabilized plaque like healed plaque, chronic total occlusion, and fibrocalcific and fibrous plaques [7]. Stary wrote editorial review in which he modified the AHA classification but still did not acknowledge the existence of plaque erosion and calcified nodule [8].
Intimal Thickening and Fatty Streaks

Development of human coronary atherosclerosis. The two nonprogressive lesions are intimal thickening or intimal xanthoma (foam cell collections known as fatty streaks, AHA Type II). Pathological intimal thickening (AHA Type III, transitional lesions) is the first of progressive plaques marked by an acellular lipid pool rich in proteoglycan; inflammation when present is typically confined to the most luminal aspect of this plaque. Fibroatheromas are lesions with areas of necrosis characterized by cellular debris and cholesterol monohydrate with varying degrees of calcification or hemorrhage. Finally, thin-cap fibroatheroma or vulnerable plaques are recognized by their relatively large necrotic cores and thin fibrous caps. (Reproduced with permission from Virmani et al. [2])
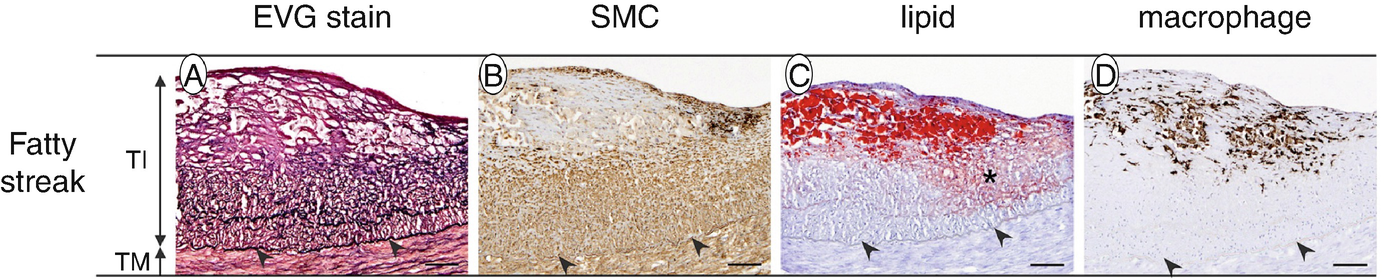
Histology of fatty streak consists of foamy macrophage infiltration of the intima with variable smooth muscle cells and proteoglycan-collagen matrix deposition. (a) EVG stain: elastic fibers were stained in black. (b) SMC: SMCs immunostained with anti-α-SMA/HHF35. (c) Lipid: extracellular and intracellular lipids stained with Sudan IV stain. Extracellular lipids in the fatty streak are indicated by an asterisk. (d) Macrophage: macrophages immunostained with an anti-CD68 antibody. Arrowheads indicate internal elastic lamina. Bars represent 100 μm. TI, tunica intima; TM, tunica media. (Reprinted from Nakagawa et al. [14], with permission from Elsevier)
Pathologic Intimal Thickening
Pathological intimal thickening (PIT, AHA Type III lesion) is considered as the earliest lesion of progressive atherosclerosis which consists of SMCs within extracellular matrix composed of proteoglycan within the lipid pool, which is located near the media [16] (Fig. 11.1). There is a relative absence of viable SMCs within the area of the lipid pool. The extracellular matrix consists of hyaluronan and proteoglycans such as versican along with lipid deposition. The PIT lesion also shows the presence of proteoglycans biglycan and decorin surrounding the SMCs. The SMCs in the intima in the region of the lipid pool undergo apoptosis near the media, and in the late phases, there is loss of SMC and accumulation of basement membrane (BM) as demonstrated by periodic acid-Schiff (PAS) stain and by transmission electron microscopy. Structural changes in the glycosaminoglycan chain of proteoglycans, i.e., versican and biglycan, are the initial proatherogenic proteoglycans which bind the atherogenic lipoproteins [17, 18]. On the other hand, enrichment in decorin in the neointima of the internal thoracic artery is observed which may play a role that results in the resistance to the development of atherosclerosis [19]. As PIT lesions become more advanced, macrophages begin to accumulate toward the lumen with eventual infiltration into the lipid pool converting the latter to a necrotic core. PIT lesions with macrophages are considered as a more advanced lesion. Specific protein expressed in the lipid pools may initiate the macrophages accumulate in PIT lesions, although the specific proteins remain unknown.
Free cholesterol is seen as fine crystalline structures (devoid of lipids) in paraffin-embedded sections within the area of lipid pool to varying degree; however, they are never excessive. The origin of free cholesterol in early phases is likely derived from dying macrophages and SMC; however the exact processes involved remain unknown [20]. Accumulation of extracellular lipid within the area of lipid pools is derived from the circulating plasma lipoprotein [21, 22]. In some experimental studies, intrinsic phenotypic changes of smooth muscle cells were observed in the initial stage of plaque formation which may contribute to SMC apoptosis rather than intrinsic role of SMC (i.e., cell-cell interactions or the microenvironment of the plaque) [23, 24]. In the setting of hyperlipidemia (cholesterol-rich), SMCs undergo apoptosis in vitro likely through Bcl-2 and Bax genes. However, function of phagocytosis is reduced in the presence of hyperlipidemia, leading to necrosis of apoptotic SMCs and leakage of intracellular IL-1. IL-1 acts on the surrounding viable SMCs and induces them to secrete IL-6 and monocyte chemotactic protein (MCP-1) which provoke the progression of atherosclerosis [25].
Microcalcifications (≥0.5 μm and typically <15 μm) are another feature in the PIT lesions, which can be identified by anionic stains such as von Kossa’s or alizarin red stains. It is likely that microcalcifications coalesce and form fragmented calcification; this may also involve collagen which acts as a scaffold, enhancing aggregation of microvesicles. The mechanisms that lead to microcalcification remain controversial with one group in favoring microvesicles as the mechanism of calcification and on the other related to apoptosis as the underlying mechanism with late coalescence. Most calcification in the early stages of PIT is microcalcification; however, fragmented calcification may be observed in a minority of cases.
Fibroatheroma
Fibroatheroma is the more advanced lesion of atherosclerosis and is distinguished from PIT lesions by the presence of an acellular necrotic core (AHA Type IV) [2] (Fig. 11.1). At this phase there is an absence or decrease in hyaluronan and proteoglycan such as versican and biglycan with the absence of collagen in the area of the necrotic core. Fibroatheromas are classified into two types, “early” and “late,” on the basis of the relative absence of matrix molecules. Early fibroatheroma show some presence of hyaluronan, versican, and biglycan and other matrix proteins due to the infiltrating macrophages within the lipid pool, which is converting to necrotic core by the focal loss of matrix. The late necrotic core is devoid of any matrix and no longer stains for hyaluronan, proteoglycans, and collagen matrix as identified by specific stains for proteoglycans, hyaluronan, and Sirius red stain. Matrix metalloproteinases (MMPs) released by macrophages are responsible for the breakdown of proteoglycan and collagen. Also, apoptotic cell death of macrophages and free apoptotic bodies are abundant within the area of necrotic core, which has been described by Ira Tabas as a “grave yard” of macrophages [26]. In addition to the deficiency of extracellular matrix (ECM), the necrotic core in late fibroatheroma exhibits more cholesterol clefts, calcification, intraplaque hemorrhage, and surrounding neoangiogenesis as compared to early fibroatheroma [27].
The most distinguishing feature of late fibroatheroma is the accumulation of free cholesterol. Acyl coenzyme A (ACAT1) esterifies free cholesterol and stores esterified cholesterol in macrophages [28]. Macrophage ACAT1 deficiency has been shown in mice to lead to an increase in atherosclerotic lesion area including free cholesterol accumulation in hyperlipidemic mice. Therefore, it is thought that ACAT1 is deficient in macrophages in the setting of advance atherosclerotic lesions. Also, accumulation of free cholesterol along with macrophage death is believed to contribute to the development toward late fibroatheroma [28, 29]. We have shown that red blood cells membranes, which are richest in free cholesterol of all cell membranes, are likely another source of free cholesterol present within the human necrotic core.
Thin-Cap Fibroatheroma
Late fibroatheromas are usually characterized by having a thick fibrous cap composed mainly of type I and III collagen, proteoglycans, and interspersed SMCs. Fibrous cap plays an important role in harboring necrotic core away from the flowing blood. As the lesion becomes more advanced, the fibrous cap can undergo thinning and is referred to as a thin-cap fibroatheroma (TCFA) although the precise mechanism still remains unknown. It is unknown whether the fibrous cap may begin as a thin cap or is converted from a thick to thin cap. The thinning of the fibrous cap is thought to result from ECM degradation by MMPs and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family, along with a deficiency in repair mechanism [30]. Although the MMPs and TIMPs in monocytes/macrophages secrete several types of extracellular proteases, which include serine proteases, cathepsins, and metalloproteinases, it is likely that overproduction of MMPs is what leads to cap thinning. The TCFA generally has large necrotic core along with intact thin fibrous cap consisting mostly of type I collagen with coexisting inflammatory cells such as macrophages and lymphocytes of varying degree. Generally, there is the absence or decrease of SMCs within the thin fibrous cap of TCFA lesions due to SMC apoptosis. We have defined thin cap as ≤65 μm based upon the thickness of ruptured cap. The ruptured caps measured 23 ± 19 μm considering 95% confidence interval of the cap; we calculated a cap thickness below ≤65 μm defining a TCFA [31]. The necrotic core was also measured and was found to be lesser in TCFA than in plaque rupture (25 ± 15% vs. 35 ± 20%, respectively) [32]. Narula et al. showed the best discriminators of TCFA besides cap thickness were macrophage infiltration and necrotic core size [32].
Plaque Rupture
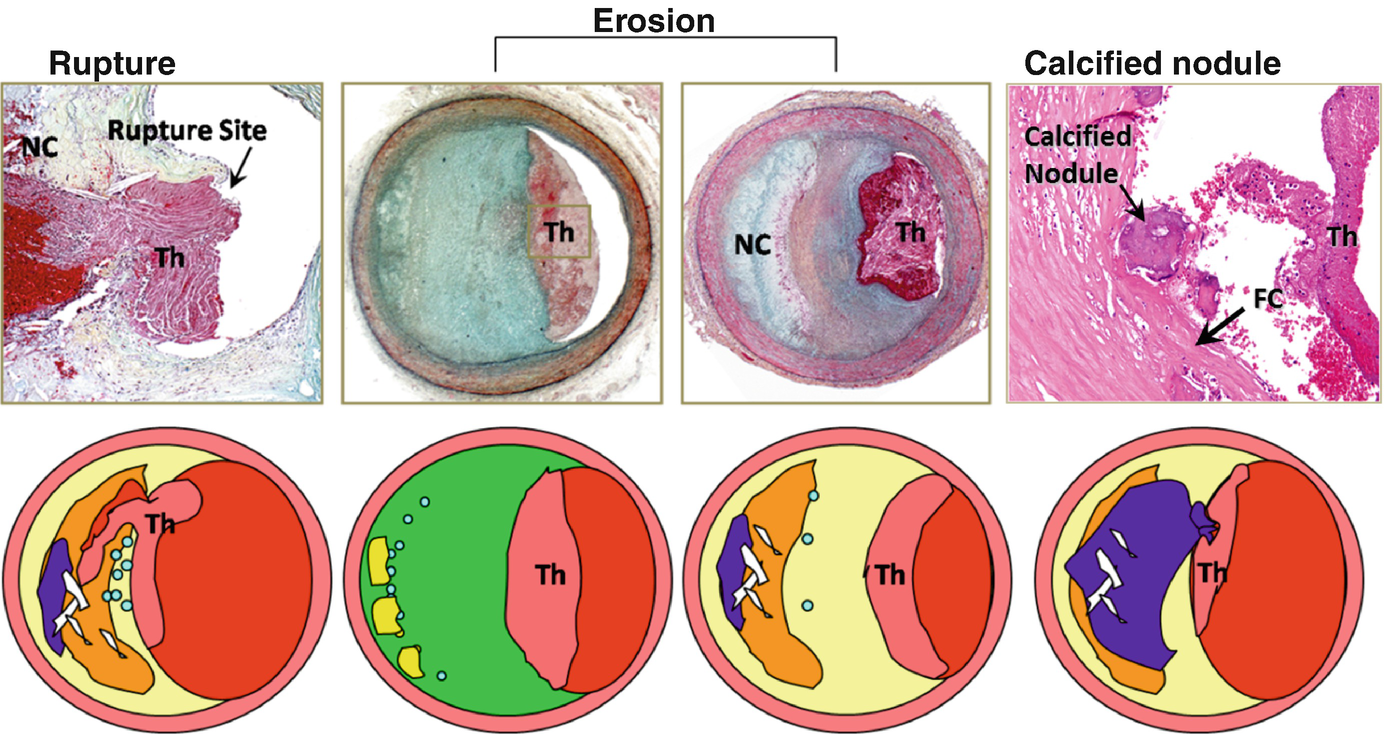
Atherosclerotic lesions with luminal thrombi. Coronary plaque features responsible for acute thrombosis comprise three different morphologies: rupture, erosion, and calcified nodules. Ruptured plaques are thin fibrous cap atheromas with luminal thrombi (Th). These lesions usually have an extensive necrotic core (NC) containing large numbers of cholesterol crystals and a thin fibrous cap (<65 μm) infiltrated by foamy macrophages and T lymphocytes. The fibrous cap is thinnest at the site of rupture and consists of a few collagen bundles and rare smooth muscle cells. The luminal thrombus is in communication with the lipid-rich necrotic core. Erosions occur over lesions rich in smooth muscle cells and proteoglycans. Luminal thrombi overlie areas lacking surface endothelium. The deep intima of the eroded plaque often shows extracellular lipid pools, but necrotic cores are uncommon; when present, the necrotic core does not communicate with the luminal thrombus. Inflammatory infiltrate is usually absent but, if present, is sparse and consists of macrophages and lymphocytes. Calcified nodules are plaques with luminal thrombi showing calcific nodules protruding into the lumen through a disrupted thin fibrous cap. There is absence of an endothelium at the site of the thrombus, and inflammatory cells (macrophages and T lymphocytes) are absent. (Reproduced with permission from Virmani et al. [2])
Related posts:
 Dysfunction and Neurodegenerative Disease
Dysfunction and Neurodegenerative Disease
 of the Carotid Artery
of the Carotid Artery
 Carotid Vessel Wall Imaging and Future Directions
Carotid Vessel Wall Imaging and Future Directions
 Processing: What Is Needed and Unique for Vessel Wall Imaging?
Processing: What Is Needed and Unique for Vessel Wall Imaging?
 Aspects of Vessel Wall Imaging: 4D Flow
Aspects of Vessel Wall Imaging: 4D Flow
 Imaging Approaches and Challenges in the Assessment of the Intracranial Vasculature
Imaging Approaches and Challenges in the Assessment of the Intracranial Vasculature
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


