Abstract
Osteogenesis imperfecta (OI) is a disorder of bone characterized by hypomineralization of the skeleton and by life-long bone fragility and fracture predisposition. OI is marked by considerable clinical and genetic heterogeneity. Historically, OI has been grouped into four major types (I-IV), with OI type II defined as perinatal lethal. More recently, OI has been divided into mild, severe, and perinatal lethal types that are associated with genetic heterogeneity. Ultrasound findings in perinatal lethal OI include skeletal hypomineralization, short limbs with angulation or bowing of the long bones, and bending of the ribs. The OI phenotypic spectrum is large, and lethality results from a small chest and underlying pulmonary hypoplasia.
Keywords
osteogenesis imperfecta, fractures, hypomineralization
Introduction
Osteogenesis imperfecta (OI), a disorder of bone characterized by hypomineralization of the skeleton, was the first disorder proposed to be due to a defect in collagen. It is characterized by life-long bone fragility and fracture predisposition. Other features, depending on the type of OI, may include blue sclerae, hearing loss, hypermobility of joints, and abnormal dentition.
OI is marked by considerable clinical and genetic heterogeneity. There is a continuum of severity, ranging from perinatal lethality to nearly asymptomatic disease. Historically, OI has been grouped into four major types (I–IV). However, several further subtypes have now been described. OI is discussed here as mild, severe, and perinatal, although this last classification is arbitrary because there is a broad spectrum of phenotypic variability.
Disorder
Definition
OI is a heterogeneous group of chondrodysplasias characterized by increased bone fragility. Mild OI (known as OI type I ) is the most common form of OI and usually has a mild clinical course. Individuals have normal type I collagen but only half the normal amount. Individuals are short but not to the degree of individuals with severe deforming OI. Fractures occur, but usually in infancy and childhood rather than in utero . Fracture healing is normal, and the limbs do not appear deformed. Abnormal dentin (dentinogenesis imperfecta) may be present, and sclerae may be blue. Other ocular defects include scleromalacia, keratoconus, and retinal detachment. In contrast to perinatal lethal OI and, often, the more severe types of OI, diagnosis is frequently missed prenatally because fractures do not usually occur in utero , and the bones appear normal by ultrasound (US).
Severe deforming OI (known as OI types III and IV ) encompasses a broad clinical spectrum. Approximately 90% of these cases are inherited as autosomal dominant. However, autosomal recessive inheritance accounts for the remaining 10% of cases. Individuals are very short, the limbs are severely deformed, the thorax is abnormal, and marked kyphoscoliosis is present. Pulmonary function can be affected by the abnormal spine and thorax; pulmonary insufficiency is the major cause of death. Less severely affected individuals usually show respiratory deterioration over time, resulting in restrictive lung disease and sleep apnea. They may have associated hydrocephalus, cranial nerve palsies, motor neuron lesions, and headache. Diplopia, nystagmus, cranial nerve neuralgia, decline in motor function, urinary dysfunction, and respiratory compromise are complications of basilar invagination. Individuals have significant generalized osteopenia, and fractures occur in the upper and lower extremities and vertebral bodies, especially before puberty. In contrast to mild OI, abnormal fracture healing leads to skeletal deformity. The diagnosis may be suggested in utero by frontal bossing, bowing of the long bones, or fractures, although the diagnosis is often missed prenatally because fractures and the accompanying skeletal deformities commonly occur later in life.
Perinatal lethal OI (known as OI type II ), also known as osteogenesis imperfecta congenita, is a perinatally lethal form of OI. Most cases are the result of de novo mutations in one of two genes that encode type I procollagen. It is important to recognize that autosomal recessive forms can present prenatally as severe OI. In utero findings include fractures at various stages of healing and very short, angulated limbs; fetal hydrops is reported in the very severe forms.
Prevalence and Epidemiology
The overall incidence of OI is approximately 0.5 : 10,000 births. OI is one of the more common prenatal onset skeletal disorders.
Etiology, Pathophysiology, and Embryology
Most cases of OI that can be diagnosed perinatally result from a dominant mutation in one of the two genes that encode the chains of type I collagen, COL1A1, and COL1A2. The result is a marked decrease in collagen of both cortical and trabecular bone. Histologically, the normal bone structure is replaced by woven bone. Abundant large osteoblasts are surrounded by little extracellular matrix. Rare recessive cases of lethal and severe OI have also been described, which are associated with mutations in genes controlling posttranslational modification and transport of collagen, including CRTAP, P3H1, PPBI, FKBP10, HSP47, SP7, PLOD2, TMEM38B, PEDF, WNT1, P4HB, SEC24D, SPRAC, SMPD3, and CREB/ATF.
Manifestations of Disease
Clinical Presentation
Rare recessive forms of perinatal lethal and severe deforming OI exist, but most cases are spontaneous autosomal dominant mutations, and diagnosis usually is made at the time of routine anatomy US, when micromelia (owing to multiple fractures) is noted. Cardinal features may be visualized by the early second trimester or sooner, particularly with transvaginal US. Perinatal lethal OI may also be associated with increased nuchal translucency in euploid fetuses. Severe deforming OI may be suggested in utero by frontal bossing, bowing of the long bones, or fractures, although the diagnosis is often missed prenatally because fractures and the accompanying skeletal deformities commonly occur later in life.
Imaging Technique and Findings
Ultrasound.
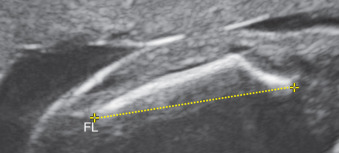
The cardinal US features of perinatal lethal OI include skeletal hypomineralization, short limbs with angulation or bowing of the long bones ( Fig. 52.1 ), and bending of the ribs ( Fig. 52.2 ). The bones may appear “wrinkled” because of multiple fractures and subsequent callus formation. The chest is small, and platyspondylisis may be apparent in the spine. Poor mineralization of the calvaria often enhances clarity of the intracranial structures ( Fig. 52.3 ) may be compressible with the US transducer ( Fig. 52.4 ). Finally, fetal movements and limb position may be abnormal.