Abstract
The type II collagen disorders are a phenotypic spectrum of diseases that include achondrogenesis II, hypochondrogenesis, platyspondylic Torrance type, spondyloepiphyseal dysplasia congenita, Kniest dysplasia, spondyloepimetaphyseal dysplasia, Strudwick type, Legg-Calve-Perthes disease, spondyloperipheral dysplasia, spondyloepiphyseal dysplasia with metatarsal shortening (Czech type), autosomal-dominant spondyloarthropathy, and Stickler syndrome. Achondrogenesis II and hypochondrogenesis can present in the first trimester with an increased nuchal fold and frequently cystic hygromas. Later in gestation, ultrasound findings, include flattened facies, small chest, short ribs with cupping, and severe shortening of all the long bones, though with relatively normal appearing hands.
Keywords
achondrogenesis II, hypochondrogenesis, Stickler syndrome, type II collagen
Introduction
Achondrogenesis II, hypochondrogenesis, platyspondylic Torrance type, spondyloepiphyseal dysplasia congenita (SEDC) ( Chapter 56 ), Kniest dysplasia, spondyloepimetaphyseal dysplasia, Strudwick type, Legg-Calve-Perthes disease, spondyloperipheral dysplasia, spondyloepiphyseal dysplasia with metatarsal shortening (Czech type), autosomal dominant spondyloarthropathy, and Stickler syndrome form the type II collagen disorder group. This group is a continuous spectrum of disorders, some of which manifest in the prenatal period, some much later in life.
Disorder
Definition
The aforementioned disorders all result from autosomal dominantly inherited disorders in the gene, COL2A1 , the gene that encodes type II collagen. Beyond sharing a common genetic basis of disease, they share some radiographic features and clinical features such as early-onset osteoarthritis in nonlethal disorders.
Prevalence and Epidemiology
The precise prevalence of this spectrum of disorders is not known but, as a group, type II collagen disorders are not uncommon among the skeletal disorders. For the lethal achondrogenesis II, the disorder is estimated to have a prevalence of 1 : 40,000 to 1 : 60,000.
Etiology and Pathophysiology
The aforementioned disorders all result from heterozygosity for mutations in the gene that encodes type II collagen, COL2A1 . The COL2A1 gene product is the type II collagen molecule. Somatic and germline mosaicism has been reported for type II collagen disorders. The type II collagen molecule provides structure and strength to connective tissues, particularly cartilage. Cartilage primarily compromises the skeleton during early development and abnormalities in type II collagen can have a profound effect on the patterning and integrity of the skeleton. Type II collagen is also part of the vitreous of the eye, the inner ear, and the nucleus pulposus; thus abnormalities in type II collagen affect the eye, the spine, and hearing.
Most of the mutations that produce these disorders result from heterozygosity for missense mutations in COL2A1 , and most of these mutations result from substitution of a glycine residue in the triple helix of the gene with a bulkier amino acid. Stickler syndrome, which presents with Pierre Robin sequence, mild short stature, and significant myopia and retinal abnormalities, results from heterozygosity for nonsense mutations in COL2A1 .
Manifestations of Disease
Clinical Presentation
The following disorders can present in the prenatal period or immediate newborn period:
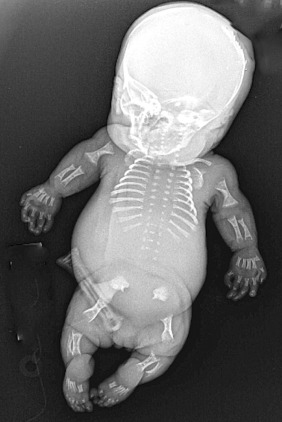
Achondrogenesis II. Achondrogenesis type II is also referred to as the Langer-Saldino type. This very rare disorder is characterized by extremely shortened arms and legs, a narrow chest with short ribs, hypoplastic lungs, and a lack of normal bone formation (ossification) in the spine and pelvis ( Fig. 57.1 ). Distinctive facial features include a prominent forehead, micrognathia, and cleft palate. The abdomen is relatively enlarged, and often these fetuses present with hydrops fetalis. Newborns with achondrogenesis type II usually die before or soon after birth.
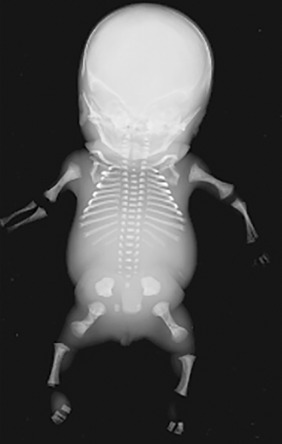
Hypochondrogenesis is very similar to achondrogenesis type II. Affected infants have short arms and legs, a small chest with short ribs, and underdeveloped lungs ( Fig. 57.2 ). Bones in the skull develop normally, but the bones of the vertebrae and pelvis lack proper ossification. Their facies appears flat and oval-shaped, with micrognathia and cleft palate (Pierre Robin sequence) and a high incidence of hydrops fetalis. Most of these infants do not survive to term. For those who do, and survive the newborn period, they are often reclassified as spondyloepiphyseal dysplasia congenita, a relatively similar but milder disorder (see Chapter 56 ).
Stickler syndrome results from heterozygosity for loss of function mutations in COL2A1 and thus is somewhat mechanistically different. These individuals may present in the prenatal period with mild shortening of the rhizomelic segment and significant micrognathia. Long-term, most people with Stickler syndrome have skeletal abnormalities that affect the joints including scoliosis and kyphosis. As children, they may have loose joints, but as they age, their joints become less flexible. Arthritis often appears early in life causing joint pain or stiffness. Of major concern are the eye findings in Stickler syndrome that include glaucoma, cataracts, and retinal detachments, increasing the likelihood of impaired vision and in some cases blindness. Sensorineural hearing loss varies in degree of severity, but is common.
Imaging Technique and Findings
Ultrasound.
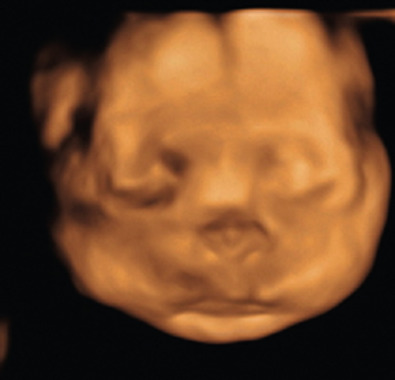
Achondrogenesis II and hypochondrogenesis will present in the prenatal period. Achondrogenesis II presents in the first trimester with an increased nuchal fold and frequently a cystic hygroma. Hypochondrogenesis will present in a similar fashion. The ultrasound findings include a flattened facies ( Fig. 57.3 ), small chest, particularly on sagittal view with short ribs with cupping at the ends ( Fig. 57.4 ), lack of secondary ossification centers, and severe shortening of all the long bones, with relatively normal-looking hands. Ascites, hydrops fetalis, and polyhydramnios are relatively common findings.