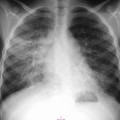
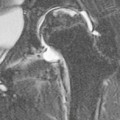
Anne Paterson, Øystein E. Olsen, Lil-Sofie Ording Müller Paediatric gastrointestinal (GI) radiology is most appropriately thought of according to the age of the patient, and for this reason the chapter has been subdivided into sections; the first details neonatal pathology, the latter relates to older children. The clinical presentation of the conditions remains as before. In this latest edition, newer embryological theories, imaging techniques, management details and updated references have been incorporated. As most children with neonatal GI problems now survive to adolescence or adulthood, data have emerged about adult cohorts and some of the longer-term health problems they face. Information detailing some of these issues has also been included. The ventral wall of the embryo is formed during the fourth week of intrauterine development as the cephalic, caudal and lateral edges of the flat, trilaminar embryonal disc fold in upon themselves and the layers fuse together. The resultant embryo is cylindrical in shape, and protruding centrally from its ventral surface are the remains of the yolk sac connected to the midgut via the omphalomesenteric duct, a structure, which normally regresses in the fifth gestational month.1,2 If this complex process is incomplete, then several types of anterior abdominal wall defect may result: ectopia cordis and pentalogy of Cantrell, gastroschisis, bladder and cloacal exstrophy, and omphalocele. In patients with gastroschisis, there is a small defect or split in the ventral abdominal wall, classically to the right side of a normally positioned umbilicus. Gastroschisis typically occurs in the absence of other anomalies and is thought to be due either to a localised intrauterine vascular accident or to asymmetry in the lateral body wall folds with failure of fusion.1 The incidence of gastroschisis has increased worldwide over the past two decades and it has been documented to occur in clusters in some geographic regions.2,3 Mothers under the age of 20 years are at greater risk of having a child with the condition.4 Antenatal ultrasound shows bowel loops floating freely in the amniotic fluid, with the diagnosis being possible early in the second trimester. There is no covering membrane. ‘Complex gastroschisis’ as determined by the presence of intestinal atresia or stenosis, bowel perforation, volvulus or necrosis is found in 10–20% of infants.3,5,6 Exposure to amniotic fluid causes damage to the bowel; postnatally, this can result in a thick, fibrous ‘peel’ coating the loops of bowel. Short bowel syndrome, liver disease secondary to intestinal failure and intestinal dysmotility are serious consequences of gastroschisis. Necrotising enterocolitis (NEC) is reported in up to 20% of patients with gastroschisis.7 Upper GI contrast studies in infants with repaired gastroschisis will often demonstrate gastro-oesophageal reflux (GOR), malrotation, dilatation of small bowel loops and a markedly prolonged transit time. Repair of the defect may be possible soon after delivery. If not, the bowel is protected in a silo to prevent fluid loss, and gradually returned to the abdominal cavity. An omphalocele (syn. exomphalos) is a midline anterior abdominal wall defect through which the solid abdominal viscera and/or bowel may herniate. The extruded abdominal contents are covered in a sac. Larger omphaloceles containing liver tissue may be due to failure of fusion of the lateral body folds. Omphaloceles containing only bowel are thought to arise due to persistence of the physiological herniation of gut after the tenth week of fetal development. The umbilical cord inserts at the tip of the defect. A giant omphalocele is said to be present when the liver is contained within the herniated membranes or when the defect measures more than 5 cm in diameter.8 Antenatal ultrasound can detect an omphalocele from the second trimester onwards. The prognosis of the infant is dependent upon associated anomalies. Chromosomal and structural abnormalities are seen in more than 50% of patients.3 The Beckwith–Wiedemann syndrome has an omphalocele (exomphalos), macroglossia and gigantism as its primary components (the ‘EMG’ syndrome). The method of surgical closure of the defect is in part driven by its size. Immediate closure, staged procedures or delayed repair following epithelialisation are all surgical possibities. The terminology of this spectrum of complex disorders is confusing, with many texts using the term OEIS complex (omphalocele, exstrophy, imperforate anus and spinal abnormality) and cloacal exstrophy, interchangeably. In reality, they probably represent different entities of the same spectrum. The epispadias–exstrophy spectrum is rare and complex, extending from epispadias, where males have a urethral meatus opening on the dorsum of the penis, and affected females a cleft urethra, to bladder exstrophy, where the bladder is exposed on the lower abdominal wall and drips urine constantly. At the far end of the spectrum is cloacal exstrophy, one of the most severe congenital anomalies compatible with life, and which encompasses abnormalities of the genitourinary (GU) and GI tracts, the central nervous and musculoskeletal systems. This condition is thought to arise due to abnormal development of the cloacal membrane9 and its premature rupture prior to the fifth week of gestation.10 The cloaca opens onto the lower abdominal wall, where it is seen as an open caecum and prolapsing terminal ileum between two hemibladders. There is an omphalocele of varying size and a blind-ending short gut. The external genitalia are ambiguous. Bilateral inguinal herniae are common to both sexes. There is spinal dysraphism and there is an ‘open book’ pelvis.11 Associated renal and lower limb anomalies (club foot and reduction defects) are well described.9 Antenatally, the ‘elephant’s trunk’ sign of the prolapsed terminal ileum is said to be pathognomonic for the condition on ultrasound.9 Non-visualisation of the bladder in association with an omphalocele and myelomeningocele would also be strong predictors of cloacal exstrophy. Early postnatal imaging includes the extensive use of ultrasound to evaluate the spine and brain, the GU tract and the hips. CT with 3D-VR images is helpful in planning pelvic surgery.11 Upper GI contrast studies document GI tract anatomy. Magnetic resonance imaging (MRI) of both the pelvis—assessing the genital tract and pelvic floor—and the spine are also required. Surgery in the immediate postnatal period includes diversion of the faecal stream and colostomy formation, bladder closure ± vesicostomy fashioning, omphalocele reduction and repair, and closure of any open neural tube defects. Genital tract surgeries are often postponed until later in childhood. The neonate with disease affecting the proximal GI tract often presents with respiratory symptoms. These symptoms include excess salivation, choking with feeds, coughing, cyanosis and respiratory distress. Conditions that present in this way include oesophageal atresia (OA) with or without tracheo-oesophageal fistula (TOF), laryngeal clefts, swallowing disorders, diaphragmatic hernias, vascular rings and GOR; the latter is by far the most common. The chest radiograph may show airspace disease and atelectasis, should aspiration have already occurred. OA with or without a fistulous connection to the trachea is one of the more common congenital anomalies of the GI tract. It is caused by abnormal partitioning of the foregut into separate respiratory (ventral) and GI (dorsal) components early on in the first trimester of fetal life. Five different major anomalies result (Fig. 77-1). The atretic segment of the oesophagus tends to be at the junction of its proximal and middle thirds. Occasionally an isolated TOF occurs without OA; this is the H-type fistula. Half of all children with OA and TOF have associated congenital anomalies. Features of the VACTERL spectrum (vertebral anomalies, anorectal malformation, cardiovascular malformation, tracheo-oesophageal fistula with oesophageal atresia, renal anomalies and limb defects) are especially common, with other GI malformations—notably duodenal atresia, small bowel malrotation and volvulus and a more distal congenital oesophageal stenosis often reported.12 OA is usually suspected on an antenatal ultrasound. The cardinal findings of maternal polyhydramnios and an absent stomach bubble have a positive predictive value of 50%.13 A false-negative exam may be as a result of fluid passing via a lower pouch fistula into the stomach. However, OA can be diagnosed with greater certainty by visualising the dilated, blind-ending upper oesophageal pouch and the use of cine-mode fetal MRI.13,14 The remaining group of infants present almost immediately in the postnatal period with choking, coughing, cyanosis and drooling, symptoms which are exacerbated during attempts to feed the infant. Patients with an H-type fistula are usually symptomatic from birth. Postnatally, the diagnosis is usually made on a chest radiograph, which shows an orogastric tube curled in the proximal oesophageal pouch (Fig. 77-2). The lungs may show features of an aspiration pneumonitis. The presence of gas in the abdomen implies a distal fistula. A gasless abdomen is seen with isolated OA or rarely OA with a proximal fistula. In those infants in whom a primary repair is not possible, the gap between the proximal and distal oesophageal pouches can be assessed following the formation of a feeding gastrostomy. Under fluoroscopic guidance, a Hegar dilator is inserted though the gastrostomy and passed retrogradely into the distal oesophagus. A Replogle tube is simultaneously used to delineate the superior pouch. As both tubes are radiopaque, the degree of separation between the pouches is easily visualised. Thoracic CT imaging, following simultaneous injection of air into the upper pouch, via the indwelling Replogle tube, and also via the gastrostomy, can alternatively be used.15,16 The standard, long-established diagnostic technique for an H-type fistula in most institutions remains the ‘withdrawal oesophagram’, obtained with the infant in the prone position, with a horizontal X-ray beam (or a steep oblique position with a vertical X-ray beam). The contrast is injected under pressure, via a nasogastric tube (NGT) with its tip in the distal oesophagus, as the tube is gradually pulled back under fluoroscopic guidance (Fig. 77-3). Care must be taken not to spill barium via the vocal cords into the trachea, or water-soluble contrast medium can be used. Combined bronchoscopy and oesophagoscopy should be performed if there is a high clinical index of suspicion with negative imaging. A positive contrast medium injection to outline the proximal oesophageal pouch is rarely necessary, but occasionally required to exclude a proximal pouch fistula. Small quantities (1–2 mL) of isotonic, non-ionic contrast medium should be used under fluoroscopic guidance. A proximal fistula can also be demonstrated if air is injected into the Replogle tube during a multidetector CT (MDCT) imaging.17 Given the frequent combination of OA/TOF with VACTERL components, a preoperative work-up should involve assessment of the cardiovascular system and GU tract. The immediate complications following OA and TOF repair are recurrence of the TOF, which occurs in up to 10% of patients, and anastomotic breakdown in a further 10–20% of patients. Major leaks will present early with a tension pneumothorax and require repeat surgery.18 Presentation of a recurrent fistula may be delayed for months after surgery, and the child can suffer recurrent aspiration pneumonitis and pneumonia. A transanastomotic feeding tube is positioned by most surgeons, who will request a postoperative oesophagram (water-soluble contrast media) prior to the recommencement of enteral feeds. Anastomotic strictures develop in up to 80% of patients,19 and are more likely in long gap OA, and in those who have marked postoperative GOR. Oesophageal strictures can easily, safely and repeatedly be treated by balloon catheter dilatation using fluoroscopic guidance. As many of the early repair cohorts are now in adulthood, data have emerged regarding the long-term problems patients face following OA/TOF surgery. Dysphagia of varying degrees is experienced in up to 60%; this can be severe enough to cause food impaction.19 Almost half of patients report GOR or regurgitation. Biopsy-proven Barrett’s oesophagus has been reported in patients’ following OA repair.19 Respiratory problems lasting into adulthood are common. These are generally mild, but include wheezing and reactive airways disease in 25%, choking sensations in 10% and recurrent infections in up to 30%.20,21 Vomiting is a common problem in children of all ages and its presence as a symptom does not necessarily indicate GI disease. Vomiting is common with infections in any body system, in metabolic disease, disorders of the central nervous system, and as a side effect of drugs and poisons. Neonatal non-bilious vomiting due to GI causes implies a lesion proximal to the ampulla of Vater, and is most frequently due to GOR. A full clinical history and physical examination are the most important factors in determining what imaging investigations (if any) are required. Congenital gastric obstruction is rare. It is usually due to a web or diaphragm in the antrum or pylorus. Occasionally a true atresia is present, with a fibrous cord uniting the two blind ends. Pyloric atresia is associated with epidermolysis bullosa simplex.22 The diagnosis may be suspected antenatally due to maternal polyhydramnios and a large fetal gastric bubble. Postnatally, non-bilious vomiting and upper abdominal distension are found. A plain radiograph will show a dilated stomach with no distal air. At ultrasound, webs appear as persistent, linear, echogenic structures arising from the antral or pyloric walls and extending centrally. Enteric duplication cysts are uncommon congenital anomalies of obscure aetiology. They can occur anywhere along the length of the gut but are most frequently found in the ileum. Gastric duplications account for less than 5% of cases,23,24 and are usually found on the greater curve. When located in the antropyloric region they may cause gastric outlet obstruction and present in the neonatal period with non-bilious vomiting and a palpable mass. Confirmation of the diagnosis is by ultrasound. The cysts have a layered wall, with an inner echogenic layer corresponding to the mucosa/submucosa and an outer hypoechoic layer that represents a smooth muscle layer (Fig. 77-4). This appearance is referred to as the ‘gut signature’. The contents of the cyst are usually hypoechoic, but debris is seen if there has been haemorrhage or infection has developed. Gastric mucosa takes up 99mTc-pertechnetate, which is helpful to identify the cysts when children present with GI bleeding (see the section ‘Enteric Duplication Cysts’ under ‘Abdominal Distension’). This is a rare abnormality characterised by a small, tubular, midline stomach, severe GOR and a mega-oesophagus. The patients present with recurrent vomiting, failure to thrive, signs of malnutrition and recurrent episodes of aspiration pneumonia. Associated anomalies are found in nearly all cases, and include components of the VACTERL sequence, small bowel malrotation, upper limb reduction defects and asplenia. An upper GI contrast study confirms the diagnosis. Gastric perforation is uncommon but accounts for a significant proportion of cases of neonatal pneumoperitoneum, and is a life-threatening condition. The underlying cause is unclear, but the rate of enteral feed introduction, ischaemia and increased mechanical pressure have been suggested.25,26 Iatrogenic gastric perforation is recognised after insertion of orogastric tubes. A massive pneumoperitoneum is seen on a plain radiograph of the abdomen. Bilious vomiting in the neonate may be the presenting feature of many different conditions. Sepsis, metabolic upset and gastroenteritis are examples requiring ‘medical management’, whereas, from a surgical perspective, bilious emesis indicates bowel obstruction distal to the ampulla of Vater. The most urgent of all emergencies in a neonate with bilious vomiting is small bowel malrotation complicated by volvulus. A plain abdominal radiograph alone will not distinguish between the plethora of underlying conditions causing bilious vomiting. However, a complete high intestinal obstruction (mid-ileal level and above), denoted by a few dilated bowel loops only, requires operative intervention irrespective of the cause, and no further imaging is necessary. If there is a low intestinal obstruction (distal ileum and beyond), as demonstrated by the presence of multiple dilated bowel loops on the plain radiograph, a single-contrast enema will generally be performed prior to any definitive treatment. Malrotation is a generic term used to describe any variation in the normal position of the intestines. In itself, it is not necessarily symptomatic, but the resultant abnormal position of the duodenojejunal flexure (DJF) ± caecum, mean these two organs lie closer together, shortening the base of the small bowel mesentery (Fig. 77-5). The midgut has a propensity to twist around this narrowed pedicle (volvulus), compromising the arterial inflow and venous drainage of the superior mesenteric vessels, which can lead to ischaemic necrosis of the small bowel. Untreated small bowel volvulus has a high mortality rate. In fetal life, the gut begins as a straight, midline tube, which, as it elongates and develops, herniates into the base of the umbilical cord. Traditional embryological teaching is based upon the theory that between the sixth and tenth weeks of fetal development, the midgut loop undergoes a complex three-stage anticlockwise rotation process centred around the axis of the superior mesenteric artery (SMA). This is said to explain the position of the DJF in the left upper, and the ileocaecal junction in the right lower quadrants of the abdomen. The mesentery of the small bowel extends between these two fixed points, giving it a broad base. More recent work by Metzger et al.27 proposes that the anatomical position of the gut relies critically upon localised growth and lengthening of the duodenal loop, which pushes it beneath the mesenteric root. Should the embryological development of the bowel be interrupted, then a variety of abnormal gut positions can occur; infants with congenital diaphragmatic herniae, gastroschisis and omphalocele by definition all have malrotated, malfixated bowel, although volvulus in these patients is rare after repair of the primary abnormality.28 In general, small bowel malrotation is an isolated abnormality, though it has been reported in association with pyloric stenosis, duodenal stenosis, web and atresia, preduodenal portal vein, annular pancreas and jejuno-ileal atresias. The heterotaxy syndromes, Hirschsprung’s disease and megacystis–microcolon–intestinal hypoperistalsis syndrome are also associated with malrotation and volvulus, as are cloacal exstrophy, prune-belly syndrome and intestinal neuronal dysplasia. Symptomatic babies with malrotation commonly present within the first month of life, with bilious vomiting. Older children may present with non-specific symptoms of chronic or intermittent abdominal pain, emesis, diarrhoea or failure to thrive. Volvulus, though less common in the older child, still occurs. It is important to suspect malrotation and volvulus in a child of any age with bilious vomiting, and to investigate accordingly in an emergent fashion. There are no specific plain radiographic findings in malrotation, even with volvulus. The radiograph may be completely normal if the volvulus is intermittent or if there is incomplete duodenal obstruction, due to a loose twisting of the bowel. If the volvulus is tight, then complete duodenal obstruction results, with gaseous distension of the stomach and proximal duodenum. The classical picture is of a partial duodenal obstruction, with distension of the stomach and proximal duodenum, with some distal gas (Fig. 77-6). A pattern of distal small bowel obstruction is seen in a closed loop obstruction and represents a more ominous finding; the small bowel loops may be thick-walled and oedematous, with pneumatosis being evident. These findings represent small bowel necrosis. A gasless abdomen is seen if vomiting has been prolonged, and in both closed loop obstruction with viable small bowel or massive midgut necrosis. In the neonate with bilious vomiting and a complete duodenal obstruction or a seriously ill child with obvious signs of peritonism, radiographic examination should cease after the plain radiograph; and urgent surgery is indicated. In all other children with bilious vomiting and incomplete bowel obstruction, further investigation—usually in the form of an upper GI contrast study—is required. This examination is best performed with barium (single-contrast study). The intestines in malrotation are malpositioned and the purpose of the upper GI study is to locate the position of the DJF. Meticulous radiological technique is required, and care must be taken not to overfill the stomach with contrast medium, as this can obscure the position of the DJF directly or secondarily by duodenal ‘flooding’ following rapid emptying of the stomach. The operator should aim to carefully define the position of the DJF on the first pass of contrast medium through the duodenum. On a supine radiograph, the normal DJF lies to the left of the left-sided vertebral pedicles at the height of the duodenal bulb (Fig. 77-7A). When malrotation is present, the DJF is usually displaced inferiorly and to the right side (Fig. 77-7B). It is important to remember that the DJF can be displaced temporarily by a distended colon or stomach, an enlarged spleen, an indwelling naso-enteric tube or manual palpation. The ‘corkscrew’ pattern of the duodenum and jejunum spiralling around the mesenteric vessels is pathognomonic for midgut volvulus on the upper GI study, the calibre of the bowel decreasing distal to the point of partial obstruction (Fig. 77-8). If there is an abrupt cut-off to the flow of contrast in the third part of the duodenum, volvulus cannot be excluded with certainty and these infants too must proceed directly to surgery. Malfixation of the intestines invariably accompanies malrotation in an attempt to fix the gut in place. Peritoneal (Ladd’s) bands stretch from the (sometimes high-lying) caecum and right colon, across the duodenum to the right upper quadrant and retroperitoneum. The Ladd’s bands themselves can cause duodenal obstruction. Ultrasound may demonstrate the dilated, fluid-filled stomach and proximal duodenum when obstruction is present. The relationship of the superior mesenteric vein (SMV) to the superior mesenteric artery (SMA) is abnormal in about two-thirds of patients with malrotation, when the vein lies ventral or to the left of the artery, a finding that is neither sensitive nor specific for malrotation. Ultrasound has also been proposed to confirm the (normal) retromesenteric position of the third part of the duodenum, and has been advocated as a screening tool in neonatal populations.29 A volvulus may be demonstrated with ultrasound as the ‘whirlpool sign’; colour Doppler studies show the SMV spiralling clockwise around the SMA. The standard operation for small bowel malrotation and volvulus is Ladd’s procedure, whereby the bowel is exposed, untwisted and inspected. Any Ladd’s bands are divided, and the base of the mesentery is widened. Finally, the bowel is returned to the abdomen, with the duodenum and jejunum to the right side, and the colon to the left; and a prophylactic appendicectomy is often performed. In those patients with small bowel malrotation diagnosed incidentally on an upper GI contrast study performed for another indication, management is less certain. Many paediatric surgeons will opt to perform an elective Ladd’s procedure. It is important that radiologists understand that a Ladd’s procedure does not result in a normal anatomical position of the bowel; the DJF will always be malpositioned in these patients and recurrent volvulus can occur if the mesenteric base is not sufficiently widened at the time of the first surgery. Duodenal atresia (DA) is much more common than duodenal stenosis, and both are caused by failure of recanalisation of the duodenal lumen after the sixth week of fetal life. Duodenal obstruction may also be caused by webs or diaphragms. Extrinsic duodenal compression by an annular pancreas or preduodenal portal vein may contribute to the obstruction in some patients. Regardless of the cause, in 80% of cases, the level of the obstruction is just distal to the ampulla of Vater. Associated anomalies are present in the majority of patients with DA or stenosis. Down’s syndrome is present in 30% of patients and congenital heart disease in 20%. Malrotation is present in 20–30% of patients and can only be diagnosed prior to surgery if the duodenal obstruction is partial. Components of the VACTERL association may also be present, with OA coexisting in up to 10% of infants. DA may be diagnosed antenatally when the dilated stomach and duodenal cap are seen. Fetal growth retardation, maternal polyhydramnios and consequent prematurity are common. Infants otherwise present early in the postnatal period with bilious vomiting and upper abdominal distension. Non-bilious vomiting occurs in those infants with a preampullary obstruction. Radiographs show a gas-filled ‘double bubble’ of the stomach and duodenal cap (Fig. 77-9). If the obstruction is partial or in the rare cases of a bifid common bile duct straddling the atretic segment, then distal gas will be present. In an upper GI study, duodenal stenosis is seen as a narrowed area in the second part of the duodenum, A duodenal web may be seen as a thin, filling defect extending across the duodenal lumen. Ultrasound can also be used to make the diagnosis, the examination made easier if the infant is first given clear fluids orally. The treatment of both complete and incomplete duodenal obstruction is surgical. Jejunal and ileal atresias have a common aetiology and are thought to be due to an intrauterine vascular accident. The vascular insult may be a primary or secondary event (for example due to antenatal volvulus or intussusception). Jejunoileal atresias have an increased incidence in patients with gastroschisis and meconium ileus. The ‘apple peel’ syndrome is thought to follow intrauterine occlusion of the distal SMA. There is a proximal jejunal atresia, with agenesis of the mesentery and absence of the mid-small bowel. The distal ileum spirals around its narrow vascular pedicle, an appearance which gives the syndrome its name. A malrotated microcolon is also usually present. A second, more complex type of intestinal atresia is the syndrome of multiple intestinal atresias with intraluminal calcifications.30 The majority of infants with a small bowel atresia present with bilious vomiting in the immediate postnatal period. With more distal atresias, abdominal distension and failure to pass meconium are more commonly recognised clinical features. On the plain radiograph, there are dilated loops of small bowel down to the level of the atresia. The loop of bowel immediately proximal to the atresia may be disproportionately dilated and have a bulbous contour. A meconium peritonitis with calcification of the peritoneum will be present if an intrauterine perforation has occurred. Management is surgical, with bowel resection and primary anastomosis if possible. In infants with multiple atresias, the surgeon aims to preserve as much of the bowel length as is feasible. Abdominal distension in the neonate may be due to mechanical or functional bowel obstruction, an abdominal mass lesion (Table 77-1
Paediatric Abdominal Imaging
Introduction
The Neonate
Visible Abnormalities of the Anterior Abdominal Wall
Gastroschisis
Omphalocele
Bladder Exstrophy—Epispadias—Cloacal Exstrophy Complex
Respiratory Distress and Choking
Oesophageal Atresia and Tracheo-Oesophageal Fistula

Early Post-Surgical Radiology.
Longer-Term Problems.
Non-bilious Vomiting
Obstruction of the Stomach
Enteric Duplication Cysts
Microgastria
Gastric Perforation
Bilious Vomiting
Small Bowel Malrotation and Volvulus
Management
Duodenal Atresia and Stenosis
Small Bowel Atresia and Stenosis
Abdominal Distension
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Paediatric Abdominal Imaging
Chapter 77