Fig. 16.1
FDG-PET scan showing serial axial images and surface-rendered z-score maps of significant hypometabolism relative to a normal control database showing selective mesial occipital hypometabolism in a patient with DLB. Additional temporoparietal and mild frontal association cortical hypometabolism is typical both of DLB and of AD
The consistent observation of a metabolic reduction in the medial occipital cortex in Parkinson dementia suggests the use of functional brain imaging as a potential clinical diagnostic aid to differentiate DLB from AD (Minoshima et al. 2001). For example, Minoshima et al. found that the presence of occipital hypometabolism distinguished DLB from AD with 90 % sensitivity and 80 % specificity in a study using a postmortem diagnostic validation (Minoshima et al. 2001). The sensitivity in discriminating DLB and AD using FDG-PET was greater than that with clinical diagnostic criteria applied retrospectively to the data from medical chart review (Luis et al. 1999; Minoshima et al. 2001).
Longitudinal studies in initially non-demented PD subjects offer an opportunity to study the metabolic pattern of incident dementia. We recently reported the results of a prospective cohort study where we compared changes in regional cerebral glucose metabolism in patients with incident dementia after 2–6 years of follow-up compared to patients who remained non-demented (Bohnen et al. 2011). We found that incident dementia in idiopathic PD initially presented as a predominant hypometabolic pathology in the visual association cortex (Brodmann area 18) and the posterior cingulum and precuneus. Although hypometabolism of the cuneus and precuneus remained most prominent, progression of dementia was associated with mixed subcortical and widespread cortical changes that also involve the mesiofrontal lobes. The anterior cingulate cortex, which has distinct connections and behavioral attributes from those of the posterior cingulated, remained relatively spared. The PDD converter patients also demonstrated relative sparing of the primary sensorimotor cortex, a pattern similar to AD and DLB. Isolated occipital glucose hypometabolism is not specific for Parkinson dementia and can also be seen in non-demented PD patients (Bohnen et al. 1999).
Resting state glucose metabolic imaging studies have been analyzed using voxel-based principal component analysis to characterize metabolic brain networks underlying cognitive dysfunction in PD (Huang et al. 2007). A specific factor identified as a PD-related cognitive spatial covariance pattern (PDCP) is characterized by metabolic reductions in frontal and parietal association areas and relative increases in the cerebellar vermis and dentate nuclei. The PDCP can be quantitatively expressed as a numerical network score in individual subjects and will allow quantitative comparison of FDG-PET scan findings over time to track progression of cognitive disease or response to treatment in PD (Mattis et al. 2011).
16.3 Dopaminergic PET Imaging in Parkinson Dementia
The basal ganglia and the neurotransmitter dopamine (DA) have been key targets for research exploring the pathophysiology underlying PD and its associated cognitive impairment. Several radiotracers have been employed previously to image nigrostriatal DA nerve terminals, including ligands metabolized by dopamine-synthetic enzymes such as [F-18]fluorodopa (FDOPA), synaptic vesicular monoamine transporter type 2 (VMAT2) ligands such as [C-11]dihydrotetrabenazine (DTBZ), and dopamine plasmalemmal DA reuptake transporter ligands such as the cocaine analogue [C-11]WIN 35,428 (Frey et al. 1996; Frost et al. 1993; Garnett et al. 1978). The caudate nucleus has more prominent non-motor, cognitive, and affective connections, whereas the putamen plays a more direct role in motor functions (Alexander et al. 1986). Dopaminergic PET studies have shown prominent reductions of putaminal greater than caudate nucleus activity in PD (Fig. 16.2) (Frey et al. 1996).
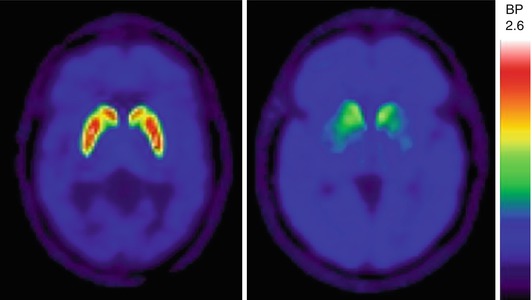
Fig. 16.2
VMAT2 PET transaxial slices showing normal striatal uptake (left) and typical nigrostriatal dopaminergic denervation pattern in a PD patient (right). The PD patient manifests left greater than right putaminal VMAT2 losses with less severe reduction of the caudate nuclei
Dopaminergic mesolimbic and mesocortical pathways that innervate parts of the limbic system and the mesiofrontal neocortex (Bjorklund and Lindvall 1984) may affect cognitive functions in PD (Kovari et al. 2003; Williams-Gray et al. 2009). A FDOPA study found relative differences in FDOPA uptake in the anterior cingulum, ventral striatum, and right caudate nucleus in PDD compared to non-demented PD patients using voxel-based analysis (Holthoff et al. 1994; Ito et al. 2002; Nagano-Saito et al. 2004). These data suggest that dementia in PD is associated with impaired mesolimbic and caudate dopaminergic function. However, a more recent FDOPA study also found evidence of significant reductions in the caudate nuclei, anterior cingulate, and mesolimbic and mesofrontal cortices in non-demented PD patients compared to normal control subjects (Klein et al. 2010). Furthermore, these authors did not find significant differences in striatal or extra-striatal cortical FDOPA activity between non-demented PD and Parkinson dementia patients (Klein et al. 2010). Other FDOPA-PET studies have also failed to find differences in striatal activity between PDD and non-demented PD patients (Hilker et al. 2005). Similar findings were reported in a recent [F-18]FP-CIT dopamine transporter PET study, where patients with PDD did not differ from non-demented PD patients in caudate nucleus or putaminal activity (Song et al. 2013). Furthermore, a longitudinal dopamine transporter [F-18]FP-CIT study found progressive decreases in striatal dopaminergic activity that correlated with changes in metabolic motor network and clinical motor scores but not with metabolic cognitive network changes in PD (Huang et al. 2007). Therefore, caudate nucleus, limbic, and mesofrontal dopaminergic denervations alone do not appear sufficient for the development of full-blown dementia in PD (Klein et al. 2010). With respect to PDD versus DLB, Klein et al. did not find significant differences in striatal, limbic, or mesofrontal FDOPA uptake between the two clinical subtypes of PD dementia (Klein et al. 2010). Patients with DLB have comparable VMAT2 nigrostriatal losses compared to PD, but there is no evidence of nigrostriatal denervation in AD (Villemagne et al. 2011, 2012). Therefore, findings of occipital hypometabolism on FDG-PET or of presynaptic nigrostriatal DA deficits can be used to distinguish PDD and DLB from AD (Burke et al. 2011; Hu et al. 2000; Koeppe et al. 2005; Villemagne et al. 2011).
16.4 Cholinergic PET Imaging in Parkinson Dementia
Parkinson disease is viewed traditionally as a motor syndrome secondary to nigrostriatal dopaminergic denervation. However, some motor and non-motor impairments may result from an intricate interplay of multisystem degenerations and neurotransmitter deficiencies that extend beyond the loss of dopaminergic nigral neurons. There is increasing neuropsychological, imaging, and pathologic evidence that suggests a common denominator referable to early dopamine deficiency but highlights an increasingly important focus on non-dopaminergic (e.g., cholinergic pathway) involvement in PD and incident dementia (Marder 2010). For example, cholinergic denervation can occur early in PD, and progressive cholinergic denervation has been consistently associated with the presence of dementia in this disorder (Bohnen et al. 2003; Hilker et al. 2005; Shimada et al. 2009; Shinotoh et al. 1999). Imaging results are consistent with postmortem evidence that basal forebrain cholinergic system degeneration appears early in PD and worsens with the appearance of dementia (Ruberg et al. 1986). Therefore, impairment or degeneration of the cholinergic system plays a significant role in the progressive cognitive decline in PD (Perry et al. 1985).
There are two major sources of cholinergic projections in the brain. The nucleus basalis of Meynert/diagonal band of Broca/medial septum complex provides the principal cholinergic input to the entire cortical mantle and degenerates in PD (Mesulam and Geula 1988). The pedunculopontine nucleus, a brainstem locomotor center, provides cholinergic inputs to the basal ganglia, thalamus, cerebellum, several brainstem nuclei, and spinal cord (Heckers et al. 1992) and also degenerates in PD (Lee et al. 2000). Acetylcholinesterase (AChE)-PET imaging can assess cholinergic terminal integrity, with cortical activity reflecting basal forebrain projections and thalamic uptake reflecting pedunculopontine nucleus integrity.
Previous studies have found cognitive correlates of cortical cholinergic activity in PD (Bohnen et al. 2006; Shimada et al. 2009), whereas subcortical (pedunculopontine nucleus-thalamic) activity has been associated with a history of falls and symptoms of REM sleep behavior disorder (Bohnen et al. 2009; Gilman et al. 2010; Kotagal et al. 2012). Therefore, cholinergic system degeneration may provide a conceptual framework to explain why PD patients with greater postural instability and gait disturbances are at an increased risk of developing dementia (Alves et al. 2006; Taylor et al. 2008). We also found that low range cholinergic activity in the thalamus, more robustly than the neocortex, is associated with symptoms of REM sleep behavior disorder (Kotagal et al. 2012). Cholinergic hypofunction may, thus, provide a possible mechanism to explain the reported link between presence of REM sleep behavior disorder and risk of dementia in PD (Marion et al. 2008). In contrast to the degeneration of the basal forebrain complex in AD, the cholinergic innervation of the striatum (mainly originating from striatal interneurons) and of the thalamus (mainly originating from the brainstem) appears to remain relatively intact in AD (Mesulam 2004). The more selective cholinergic denervation of the cerebral cortex may explain the lower frequency of REM sleep behavior disorder in AD compared to Parkinson dementia (Gagnon et al. 2006).
16.5 Fibrillary β-Amyloid PET Imaging in Parkinson Dementia
Neurodegenerative disorders are characterized by progressive dysfunction of specific populations of neurons. Neuronal loss is often associated with extra- and intracellular accumulations of misfolded proteins, typical of many neurodegenerative proteinopathies. For example, extracellular fibrillary amyloid deposits characterize AD, and intracellular inclusions, such as Lewy bodies, containing α-synuclein, are characteristic of PD (Irvine et al. 2008). Although PD is pathologically characterized by the presence of intraneuronal Lewy inclusion bodies, β-amyloid deposition can also be associated to varying degree, especially when the disease is complicated by dementia (Compta et al. 2011). The relative contributions of cortical and subcortical Aβ-amyloid deposition to cerebral dysfunction are unclear, and there are varying reports regarding the extent of β-amyloid depositions in PD versus PD dementia. Previous β-amyloid PET imaging studies using the Pittsburgh compound B (PiB) ligand have generally shown higher fibrillary β-amyloid burden in DLB compared to PDD (Edison et al. 2008; Gomperts et al. 2008; Maetzler et al. 2009; Rowe et al. 2007). For example, Johansson et al. did not find evidence of abnormal PiB retention in early-stage PD subjects (Johansson et al. 2008). Maetzler and colleagues found abnormal cortical PiB binding in two out of ten PDD subjects (Maetzler et al. 2008). Edison et al. found high global cortical amyloid burden in DLB, but not in PDD (Edison et al. 2008). In the PDD series, 2 out of 12 patients had both elevated cortical and striatal PiB retention. However, increased striatal PiB activity was also seen in two out of ten PDD patients who had no abnormal cortical retention. Interestingly, a recent postmortem study found greater frequency of striatal β-amyloid deposition in PDD compared to PD (Kalaitzakis et al. 2008), and may imply a role of intrinsic striatal pathology in PD dementia. Gomperts et al. compared cortical PiB retention in PD, PDD, DLB, AD, and control groups and found higher cortical amyloid burden in DLB, at levels comparable to AD. Amyloid deposition in the PDD group was low, comparable to the PD and NC groups (Gomperts et al. 2008). However, a study by Foster and colleagues found no major differences in the degree of cortical or caudate nucleus PiB binding among groups of PD (without or with mild cognitive impairment), PDD, or DLB (Foster et al. 2010) with the majority of subjects in each group having low level of PiB binding.
A postmortem correlation study of in vivo amyloid imaging in three PDD subjects found that two subjects had abnormal cortical amyloid binding on PET imaging (Burack et al. 2010). At autopsy, all three patients had abundant cortical Lewy bodies but were classified as low-probability AD based on NIA-Reagan criteria. The two amyloid PET-positive individuals had abundant diffuse β-amyloid plaques but only sparse neuritic plaques and intermediate neurofibrillary tangle pathology. The amyloid PET-negative subject had rare diffuse plaques, no neuritic plaques, and low neurofibrillary tangle burden. These postmortem correlates confirm that [11C]PiB-PET is specific for fibrillar β-amyloid molecular pathology but not for pathologic diagnosis of comorbid AD in patients with PDD due to the inability to distinguish between diffuse and neurotic plaques (Burack et al. 2010). Figure 16.3 shows a pattern of mildly increased cortical β-amyloid activity in a patient with PDD relative to a normal control person.
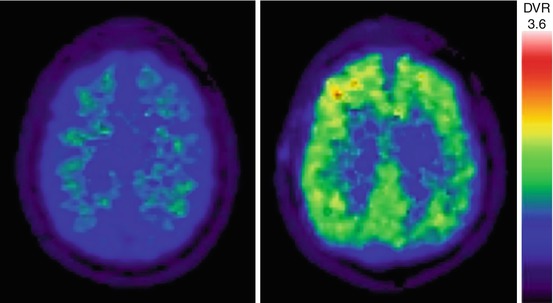
Fig. 16.3
Transaxial PiB-PET images of mildly increased cortical β-amyloid activity in a patient with PDD (right) relative to a normal control person with no abnormal β-amyloid deposition (left)
Findings of generally higher (or more frequently elevated) global cortical amyloid burden in DLB than in PDD suggest that cortical fibrillary β-amyloid deposition is not a requisite for PD dementia. However, it may differentially contribute to the temporal manifestations and/or nature of the neurobehavioral phenotype of parkinsonian dementia. Whether a combination of amyloid and Lewy body pathology exacerbates the memory and other cognitive problems in DLB is unclear, but it likely accelerates the dementia process, whereas Lewy body pathology alone may lead to a slower dementing process in PDD (Ballard et al. 2006; Petrou et al. 2012).
There are few studies that have directly correlated β-amyloid burden and cognitive performance in PD. We performed [C-11]PiB-PET imaging in 40 patients with PD at high risk for development of dementia (Petrou et al. 2012). Detailed neuropsychological testing and [C-11]DTBZ VMAT2 imaging were also performed. We found that elevated cerebral PiB binding to levels seen in patients with AD were infrequent (only 6 of 40 subjects; 15 %). Significant correlation was noted between cortical PiB binding and global composite cognitive function (r = −0.55, P < 0.005). Principal component analysis identified a factor with significant component loadings on cognitive parameters and PiB binding, but without significant striatal VMAT2 binding or other PD feature loadings. Although average cortical PiB binding in PD was in the range below those commonly used to classify PiB-PET scans as typical of AD, these apparent low binding values have significant correlation with cognitive function. Furthermore, similar relationships were observed with striatal PiB activity. These findings suggest that subcortical and cortical amyloid binding in PD and mild cognitive impairment, even at mildly elevated levels, may be of pathophysiological significance. The data also show that the lower symptomatic threshold of amyloidopathy may reflect the coexistence of other neurodegenerations, such as α-synucleinopathy, monoaminergic, and cholinergic denervations. The presence of multisystem neurodegeneration may underlie a decreased cognitive reserve capacity in PD and may explain the apparent significant cognitive correlates of even low levels of β-amyloidopathy. There is a clear need for the development of new PET ligands that are specific for neurofibrillary tau or α-synuclein proteinopathies which may help to improve our understanding of the overlap between Alzheimer pathology and dementia in parkinsonian disorders.
16.6 Multitracer PET Imaging Studies: Dopaminergic and Cholinergic Imaging Studies in PD and Parkinson Dementia
We recently reported findings of [C-11]dihydrotetrabenazine VMAT2 and AChE [C-11]PMP-PET studies in 101 patients with PD without dementia (Bohnen et al. 2012). We found that patients with lower cortical AChE activity had significantly reduced cognitive performance scores for verbal learning, executive, and attention functions compared to patients with preserved cortical cholinergic activity. Analysis of covariance showed that global cognitive functions scores had independent regression with both cortical AChE activity (F = 9.45, P = 0.0069) and nigrostriatal VMAT2 binding (F = 7.50, P = 0.0074). We did not find evidence of either exclusive cholinergic or dopaminergic substrates for specific cognitive domains to identify neurotransmitter-specific cognitive circuits. Although dopamine has long been known to modulate corticostriatal circuits and performance on executive tasks such as working memory (Goldman-Rakic 1998) and attentional control (Williams-Gray et al. 2008), we found evidence of independent cholinergic and dopaminergic effects underlying this group of functions in PD. These findings indicate that multiple neurotransmitter deficits likely contribute in parallel to cognitive impairment in PD (Kehagia et al. 2010). There are two studies that have directly compared FDOPA- and AChE-PET imaging studies in patients with PD and Parkinson dementia (Hilker et al. 2005; Klein et al. 2010). Hilker and colleagues performed MP4A AChE- and FDOPA-PET studies in 17 non-demented PD and ten PDD patients (Hilker et al. 2005). Data were compared to 31 age-matched controls. The striatal FDOPA uptake was significantly decreased in PD and PDD, without differences between these two groups. The global cortical MP4A binding was severely reduced in PDD (29.7 %, P < 0.001 versus controls) but only moderately decreased in PD (10.7 %, P < 0.01 versus controls). The PDD group had lower cortical MP4A uptake rates than did patients with PD. This group of investigators also reported on FDOPA-, MP4A-, and FDG-PET findings in eight PDD, six DLB, nine non-demented PD, and age-matched control subjects in a subsequent study (Klein et al. 2010). They reported reduced FDOPA uptake in the striatum and in limbic and associative prefrontal areas in all parkinsonian patient groups. Patients with PDD and patients with DLB showed severe MP4A and FDG reductions in the neocortex with worsening deficits from frontal to occipital regions. Significant differences between PDD and DLB were not found with any of the radioligands used. Patients with PD without dementia had mild cholinergic deficits and no significant FDG reductions versus controls. The authors concluded that patients with PDD and DLB share the same dopaminergic and cholinergic deficit profile and that cholinergic deficits seem to be crucial for the development of Parkinson dementia.
16.7 Discussion
Cognitive impairment in PD likely results from a combination of different pathologies in this pleomorphic neurodegeneration. Degeneration of subcortical systems, such as the dopaminergic nigrostriatal projection and the basal forebrain cholinergic corticopetal system, plus neurodegeneration associated with cortical depositions of α-synuclein and β-amyloid are likely mutual contributors to cognitive impairment in PD (Kehagia et al. 2010). Glucose metabolic imaging studies have shown evidence of both posterior (parieto-occipital cortical) and anterior (frontal cortical and caudate nucleus) hypometabolism in PDD (Bohnen and Albin 2011). These regional changes may reflect distinct cognitive syndromes in PD with cognitive impairment. For example, Williams-Gray and colleagues reported that more posterior cortically based cognitive defects evolved over time into dementia, whereas frontostriatal executive deficits were not associated with subsequent dementia risk per se (Williams-Gray et al. 2009). It is plausible that the glucose metabolic changes in the caudate nucleus and frontal cortex may be in part associated with dopaminergic denervation (Polito et al. 2012), whereas the more posterior cortical changes may in part be associated with cholinergic denervation (Klein et al. 2010). These data agree with dopaminergic PET studies showing that dopaminergic denervation of the caudate nucleus and fronto-limbic system alone is not sufficient for Parkinson dementia (Hilker et al. 2005; Klein et al. 2010). However, cholinergic imaging studies agree with postmortem evidence suggesting that primary basal forebrain cholinergic system degeneration significantly potentiates with the appearance of dementia. PET assessment of subcortical pedunculopontine nucleus-thalamic cholinergic denervation in PD has been associated with REM sleep behavior disorder. If basal forebrain cholinergic lesions develop in parallel, this may explain why REM sleep behavior disorder is a reported harbinger for the development of dementia in PD. Furthermore, the association between a history of falls and subcortical cholinergic denervation in PD may in part provide additional insight into why PD patients with postural instability and gait disorders also appear at higher risk of developing dementia. The comorbid presence of two neurodegenerations (α-synuclein and β-amyloidopathy) in some patients with Parkinson dementia may suggest additive or even synergistic detrimental interactions of these proteinopathies (Tsigelny et al. 2008). This may explain our findings that even low levels of cerebral amyloidopathy are significantly related to cognitive impairment in PD. These data are further consistent with postmortem findings that the presence of cortical amyloid pathology may be associated with a shorter time to development of Parkinson dementia (Compta et al. 2011). Cerebrospinal fluid amyloid β42 levels also have independent prediction of future cognitive decline in non-demented PD patients, supporting a significant role of Alzheimer pathology in the development of Parkinson dementia (Siderowf et al. 2010).
Related posts:
 Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
 PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
 Gliomas
Gliomas
 PET Reveals Pathophysiology in Ischemic Stroke
PET Reveals Pathophysiology in Ischemic Stroke
 Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
 Radionuclide Imaging Studies in Pediatric Neurology
Radionuclide Imaging Studies in Pediatric Neurology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


