Imaging procedure
Author, year
Patients
Key finding
Tc-99m-HMPAO-, Tc-99m-ECD-, I-123-IMP- or I-133-xenon- SPECT
Blend et al. (2010)
HE and carotid stenosis
Regional hypoperfusion
Catafau et al. (2000)
Subclinical HE
Regional hypo-/hyperperfusion
Ikeda et al. (2001)
Before and after liver transplantation
No change in perfusion
Liver cirrhosis
Regional hypoperfusion
Iwasa et al. (2003)
Liver cirrhosis
Hypoperfusion improved after BCAA therapy
Jalan et al. (2003)
Liver cirrhosis
Regional hypoperfusion after amino acid load
Kohira et al. (1994)
HE
Regional hyperperfusion
Nakagawa et al. (2004)
Minimal HE
Regional hypoperfusion
Ohta et al. (1998)
Portal-systemic encephalopathy
Regional hypo-/hyperperfusion improved after therapy
Strauss et al. (1999)
Fulminant hepatic failure
Reduced anterior-to-posterior perfusion ratio
Trzepacz et al. (1994)
Subclinical HE
Regional hypoperfusion
Yamamoto et al. (2011)
HE and cerebral artery aneurysm
Regional hypoperfusion
Yazgan et al. (2003)
Subclinical HE
Regional hypo-/hyperperfusion
O-15-water with/without N-13-ammonia PET
Ahl et al. (2004)
Liver cirrhosis, minimal HE
Reduced regional CBF in lenticular nucleus and cerebellar vermis, no change in PS product
Dam et al. (2013)
Liver cirrhosis, acute HE
Reduced global CBF, normalised after recovery from HE
Goldbecker et al. (2010)
Liver fibrosis, no HE
No changes in global or regional CBF and PS product
Iversen et al. (2009)
Liver cirrhosis, acute HE
Reduced global and regional CBF (frontal, parietal, temporal and occipital cortices, striatum and thalamus)
Keiding et al. (2006)
Liver cirrhosis, acute HE
Correlation between ammonia in blood and trapping in brain, no change in PS product
Lockwood et al. (1979)
Liver disease
Correlation between ammonia in blood and trapping in brain
Lockwood et al. (1991)
Minimal HE
Increased PS product
F-18-fluorodeoxy-glucose (FDG) or O-15-oxygen PET
Kato et al. (2000)
Subclinical HE
Reduced regional glucose metabolism
Giewekemeyer et al. (2007)
Early HE
Reduced regional glucose metabolism
Iversen et al. (2009)
Overt HE
Reduced regional oxygen metabolism
Senzolo et al. (2009)
Before and after liver transplantation
Improvement of glucose metabolism
C-11-PK11195 PET
Berding et al. (2010)
HCV infected
Increased marker binding
Berding et al. (2012)
Alcohol-toxic cirrhosis
No increased marker binding
Cagnin et al. (2006)
HE
Increased marker binding
Iversen et al. (2006)
HE
No increased marker binding
C-11-flumazenil PET
Jalan et al. (2000)
HE
Increased radioligand binding
MacDonald et al. (1997)
HE
Increased radioligand binding
Samson et al. (1987)
HE
Increased radioligand binding
I-123-ß-CIT or I-123-IBZM SPECT
Weissenborn et al. (2000)
HE
Reduced dopamine transporter and receptor binding
I-123-ß-CIT SPECT
Weissenborn et al. (2006)
HCV infected
Reduced dopamine and serotonin transporter binding
Heeren et al (2011)
HCV infected
F-18-CPFPX PET
Boy et al. (2008)
Liver cirrhosis
Reduced adenosine receptor binding
37.2.2 Regional Cerebral Blood Flow: SPECT and PET
Studies of brain perfusion using SPECT tracers require limited resources and provide nevertheless a robust insight into cerebrovascular changes and regional functional activity. Beyond that PET enables absolute measurements of regional cerebral blood flow (CBF), but it is technically more complex, e.g. requiring an on-site cyclotron, invasive blood sampling, etc., and higher priced. Both methods allow to assess treatment effects on the brain with an objective measure and correlate this to clinical symptoms and neuropsychological testing.
The comparability of SPECT studies on resting-state brain perfusion in patients with liver disease is sometimes limited by the fact that presence and extent of HE are not properly documented. Nevertheless, in the spectrum of patients with liver cirrhosis, if documented without HE, with minimal HE or overt HE, mostly relative regional hypoperfusion of cerebral cortex has been observed with SPECT (Catafau et al. 2000; Ohta et al. 1998; Iwasa et al. 2000, 2005; Nakagawa et al. 2004; Trzepacz et al. 1994; Yazgan et al. 2003). Some studies report (coexisting) hyperperfusion in areas of the limbic system, e.g. the basal ganglia (Catafau et al. 2000; Ohta et al. 1998; Kohira et al. 1994; Yazgan et al. 2003). Hypoperfusion involves most frequently parts of the frontal cortex (Iwasa et al. 2000; Nakagawa et al. 2004), particularly prefrontal (Catafau et al. 2000; Nakagawa et al. 2004); parietal cortex (Ohta et al. 1998; Iwasa et al. 2000; Nakagawa et al. 2004); cingulum (Nakagawa et al. 2004), particularly the anterior cingulate (Iwasa et al. 2005); and sometimes the temporal cortex (Iwasa et al. 2000; Trzepacz et al. 1994). PET studies obtaining absolute measures of cerebral blood flow in patients with liver disease substantiate the presence of flow impairments (Ahl et al. 2004; Dam et al. 2013; Goldbecker et al. 2010; Iversen et al. 2009; Keiding et al. 2006; Lockwood et al. 1991). Ahl et al. (2004) found in liver cirrhosis patients with HE compared to those without HE reduced CBF within lenticular nucleus and cerebellar vermis. Widespread reductions of regional CBF were observed in cirrhosis patients with an acute episode of HE compared to healthy subjects – encompassing areas within the frontal, parietal, temporal and occipital cortices as well as striatum and thalamus (Iversen et al. 2009). With respect to global changes of CBF in patients with liver disease, findings are consistently pointing to a decrease. Not significant decreases of 7 % in patients with liver fibrosis without HE (Goldbecker et al. 2010) and around 20 % in minimal or early HE (Ahl et al. 2004; Lockwood et al. 1991) were reported. A significant decrease of 35–40 % in global CBF was found during acute episodes of HE, reversing to an insignificant decrease of 11 % after recovery from HE (Dam et al. 2013; Iversen et al. 2009).
A positive correlation between regional cerebral perfusion assessed by SPECT imaging and scores obtained in neuropsychological testing has been reported by some authors (Catafau et al. 2000; Iwasa et al. 2005; Trzepacz et al. 1994). Results of brain perfusion measurements in general might suggest the possibility of a higher susceptibility of phylogenetically younger brain regions in particular the frontal cortex to pathophysiologic changes induced by late-stage liver disease which is in line with observed neuropsychological deficits in attention and visuopractic domains observed in neuropsychological testing (Trzepacz et al. 1994; Strauss et al. 1999).
Two interventional studies have focused on acute changes of brain (patho)physiology in patients with advanced liver disease. Jalan et al. (2003) simulated in patients with liver cirrhosis a gastrointestinal bleeding by oral administration of amino acids (mimicking haemoglobin). This resulted into a significant increase of blood ammonia levels and deterioration in neuropsychological test results. Sequential SPECT studies prior to administration of amino acids and 4 h later revealed reduced perfusion in both temporal lobes, left superior frontal gyrus, right parietal and cingulate gyrus. The study stresses the relevance of ammonia and the time course of the pathophysiology. Strauss et al. (1999) studied in patients with fulminant hepatic failure whether the use of hyperventilation to prevent or postpone the development of cerebral oedema and intracranial hypertension has an influence of cerebral perfusion. They employed xenon-133 and Tc-99-HMPAO SPECT for that purpose. No significant difference between perfusion during normoventilation vs. hyperventilation was observed. Nevertheless, perfusion was impaired in the frontal cortex of patients compared to healthy controls and was restored to normal in patients after hepatic recovery and disappearance of HE.
Effects of treatment for liver failure on HE and brain perfusion have been investigated as well. Two case report studies looked at acutely occurring HE in the situation of pre-existing cerebrovascular disease – specifically stenosis of the internal carotid artery and status after clipping of the medium cerebral artery for aneurysm, respectively (Blend et al. 2010; Yamamoto et al. 2011). In the second study of Yamamoto et al. (2011), HE was accompanied by hemispheric symptoms on neurological exam. Therapy with lactulose or branched-chain amino acids resulted into relieve from HE and hemispheric symptoms. Brain perfusion SPECT showed at baseline unilaterally hemispheric hypoperfusion in both cases and in follow-up scans after successful therapy normalisation of perfusion in one but no change in the case with initially hemispheric symptoms. The authors interpreted hypoperfusion, to reflect at baseline cerebrovascular disease and on top temporary alteration of brain tissue as a result of liver failure and in particular increased blood ammonia. Reversal of liver failure consequently resulted in normalisation of brain perfusion at least in one case.
In a study of patients with liver cirrhosis and without overt HE, treatment with branched-chain amino acids resulted in a 13–20 % increase of blood flow as demonstrated using sequential brain perfusion SPECT (Iwasa et al. 2003). PET has been used to study the impact of transjugular intrahepatic portosystemic shunting (TIPS) as a therapy option for end-stage liver disease, which is carrying a risk for the development of HE. No change of cerebral blood flow before and after TIPS was found – although flow values varied considerably preinterventionally (Iversen et al. 2011). Finally, a study of patients with citrullinaemia type II brain perfusion SPECT before and 6–36 months after liver transplantation failed to show significant changes – however, no marked decreases of perfusion had been present preoperatively (Ikeda et al. 2001).
37.2.3 Ammonia: PET
Due to the central role of ammonia in the pathophysiology of HE, N-13-labelled ammonia has been employed to further elucidate the pathogenesis. If N-13 ammonia is used in conjunction with measurements of blood flow using O-15-water, the permeability of the blood-brain barrier for ammonia can be assessed by calculation of the so-called permeability surface (PS) product (Berding et al. 2009).
This concept has been applied in the context of HE first by Lockwood et al. (1991) in patients with minimal HE. He reported a globally increased permeability supporting a key role of ammonia in HE pathogenesis and even relevance in cases with moderately or a lack of increased ammonia blood levels. This paper had a major impact on the conception of pathophysiology in HE; however, the results have not been reproduced and criticised for methodological reasons. Two points of criticism have been addressed: 1. the lack of measuring metabolites in this study might disregard radioactive metabolites entering the brain and 2. the lack of dynamic acquisition eliminated the possibility to apply full bio-kinetic modelling including consideration of backflux of tracer to the blood (Sørensen and Ott 2012).
Ahl et al. (2004) found no significant difference in global PS product between liver cirrhosis patients with and without minimal HE. However, the PS product in the thalamus was even increased in patients without HE, possibly implicating that increased permeability of the blood-brain barrier might be an early phenomenon of disease. Nevertheless, a later study of the same group comparing the PS product in patients with liver fibrosis to that in healthy controls revealed no significant difference (Goldbecker et al. 2010). Likewise, studies of liver cirrhosis patients carried out at the University Hospital Aarhus did not show significant changes of the PS product in patients without HE and those with acute overt HE during the episode as well as after recovery compared to healthy control subjects (Dam et al. 2013; Keiding et al. 2006).
Interestingly, with respect to ammonia trapping in the brain, studies comparing ammonia concentration in the blood and the metabolic rate of ammonia in the brain (derived by multiplication of net ammonia influx and arterial ammonia concentration) demonstrated a linear correlation (Keiding et al. 2006; Lockwood et al. 1979). Proceeding from the lack of specific change in blood-brain barrier permeability for ammonia shown by the above-mentioned studies using a state-of-the-art approach for quantification, this implies that the pathophysiologically relevant concentration of ammonia in the brain is simply driven by the blood ammonia concentration.
37.2.4 Energy Metabolism: PET
Measurements of energy metabolism have been employed to improve understanding of pathophysiology and therapy effects in HE. First, Kato et al. (2000) demonstrated hypometabolism in the frontal and temporal cortices as well as the basal ganglia of patients with subclinical HE compared to control subjects. A study of Giewekemeyer et al. (2007) aimed to identify brain regions relevant for bradykinesia, as an early sign of HE. They performed F-18-flurodeoxyglucose (FDG) PET studies at rest in patients with normal neurological status and either no or minimal HE according to portosystemic encephalopathy (PSE) syndrome test. Significant correlations between motor function (finger and hand tapping) and glucose metabolism could be demonstrated for brain areas relevant for movement initiation (cingulate gyrus, frontomedial, frontodorsal and parietal cortices). Iversen et al. (2009) studied oxygen consumption using oxygen-15 (O-15) PET in patients with cirrhosis without HE and with an acute episode of HE. They reported a clear reduction during the acute episode but not in patients without HE. Moreover, oxygen consumption was negatively correlated to plasma concentrations of ammonia. The authors conclude that the reduction is associated with the pathophysiology of HE and not the cirrhosis as such.
The impact of therapy for end-stage liver disease on brain energy metabolism was investigated by Senzolo et al. (2009). He studied in cirrhotic patients without overt HE but neuropsychological deficits cerebral glucose metabolism using F-18-FDG PET before and 1 year after liver transplantation. Significant beneficial effects of transplantation on initially impaired metabolism in most of the cortical (frontal, temporal, occipital) and subcortical regions corresponding with improvements in neuropsychological testing could be demonstrated.
37.2.5 Neuroinflammation: PET
In patients with hepatic disorders affecting the brain, a widely used first-generation marker of activated microglia (C-11-PK11195) has been applied. However, this marker has certain limitations, such as a relatively high non-specific uptake/low signal-to-noise ratio and complexity of quantification. Therefore, intense search for alternative markers is going on (Chauveau et al. 2008; Owen and Matthews 2011). The target for these radiotracers is the 18 kDa translocator protein (TSPO), which is present at very low levels in the normal healthy CNS but shows highly enhanced expression in the neuroinflammatory state. TSPO is located intracellular at the mitochondrial membrane. Due to TSPO presence in peripheral tissues like the kidney and high affinity to benzodiazepines, it has originally been named as peripheral benzodiazepine receptor. Its functions are not fully understood, but they include the control of cholesterol transport, neurosteroid synthesis and maybe also release of pro-inflammatory cytokines (Politis et al. 2012; Venneti et al. 2013).
Results of experimental studies imply an involvement of TSPO in the pathogenesis of encephalopathy in hepatic disease. Exposure of cultured astrocytes to ammonia results in increased TSPO expression. Exposure of mice to ammonia results into increased H-3-PK11195 binding/TSPO expression in the brain (Desjardins and Butterworth 2002). Consequently, C-11-PK11195 has been applied in patients with HE – nevertheless, results reported in the literature so far are ambiguous. Cagnin et al. (2006) observed in patients with minimal HE significantly increased C-11-PK11195 binding in the basal ganglia and the prefrontal cortex. In contrast, Iversen et al. (2006) found in patients with alcoholic liver cirrhosis during an acute episode of clinically overt HE no significant difference in C-11-PK11195 binding compared to healthy control subjects. Controversial findings might be related to different inclusion criteria, encompassing patients with hepatitis (3 out of 5) in the study of Cagnin et al. (2006), while this condition is not declared in the paper of Iversen et al. (2006). In a study employing C-11-PK11195 in a homogeneous group of hepatitis C-infected patients with fatigue, significantly increased binding was observed in the basal ganglia (thalamus) and frontal cortex (Berding et al. 2010). A preliminary study from the same group comparing liver cirrhosis patients according to the cause of liver disease, patients with cirrhosis due to hepatitis C showed increased C-11-PK11195 binding in frontal and occipital cortices, while no increase was found in those with alcoholic cirrhosis (Berding et al. 2012). Corresponding glass brain displays showing significant differences in C-11-PK11195 binding between the respective groups are included in Fig. 37.1. Thus, in summary, the presently available data suggests that neuroinflammation in the brain depends on the cause of liver cirrhosis and is specifically driven by hepatitis C virus infection.
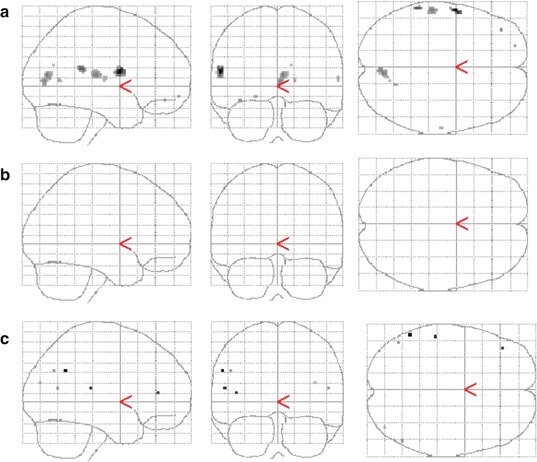
Fig. 37.1
C-11-PK11195 PET imaging of neuroinflammation in hepatic encephalopathy indicates the relevance of underlying liver disease. Displayed are the results of group comparisons with statistical parametric mapping. Regionally increased tracer binding (p < 0.001, uncorrected) is shown: in hepatitis C-infected patients vs. normal controls (a) and vs. patients with alcohol-toxic cirrhosis (c). No significant difference is seen between patients with alcohol-toxic cirrhosis and normal controls (b)
37.2.6 Neurotransmission: PET and SPECT
Specific detection of molecular processes in signal transduction between neurons is one of the exclusive goals for radiotracer studies. With respect to HE, there are hypotheses for preferential damage of specific neurotransmission systems. Particularly, the gamma-aminobutyric acid (GABA)/central benzodiazepine receptor system has been implicated to play a role in HE pathophysiology. The assumption of an increased GABAergic tone has primarily been based on the observation of beneficial effects of benzodiazepine antagonists such as flumazenil in patients with HE and animal models of HE (Albrecht and Jones 1999). Several partly controversial explanations for this increased tone have been given. A decrease in hepatic metabolism of benzodiazepines and an increase in benzodiazepine precursors provided by the intestinal flora (both resulting into increased natural benzodiazepines in the brain), an increased permeability of the blood-brain barrier for GABA and a modulations of the GABA-A receptors by ammonia or neurosteroids (Albrecht and Jones 1999; Ahboucha and Butterworth 2004; Patel et al. 2012).
Related posts:
 Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
 PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
 Single-Photon Emission Computed Tomography [Neuro-SPECT] Imaging of Brain Tumors
Single-Photon Emission Computed Tomography [Neuro-SPECT] Imaging of Brain Tumors
 PET Reveals Pathophysiology in Ischemic Stroke
PET Reveals Pathophysiology in Ischemic Stroke
 Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
 Radionuclide Imaging Studies in Pediatric Neurology
Radionuclide Imaging Studies in Pediatric Neurology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


