PET Imaging as a Biomarker
Wolfgang A. Weber
Caroline C. Sigman
Gary J. Kelloff
Biomarkers have been defined as “characteristics that are objectively measured and evaluated as indicators of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” (1). In oncology the main focus of research has been to develop biomarkers for prediction of patients’ prognosis, selection of therapeutic approaches, and assessment of tumor response to therapy. DNA chips and mass spectrometry now allow simultaneous study of several thousands of genes or proteins expressed by cancer cells that can be used as potential biomarkers. In contrast, the number of biological parameters that can be studied clinically by positron emission tomography (PET) imaging is currently limited (Table 15.1). In other words, modern genomics and proteomics provide a detailed “signature” of the cancer, whereas PET appears, at first inspection, to provide at best a few “dots.” Therefore, the obvious question is what can PET add to the huge amount of other, possible biomarkers?
PET differs in several ways from biomarkers derived from tumor biopsies or plasma samples. PET and other imaging biomarkers allow noninvasive serial studies of the whole tumor mass. In contrast, only small parts of the tumor can be evaluated by biopsies. Intratumoral heterogeneity may, therefore, significantly confound the analysis of biomarkers derived from tumor biopsies. This is of particular concern when evaluating changes in a biomarker during treatment, because it is difficult to ascertain that the expression of the biomarker in different parts of the tumor was the same prior to therapy.
Serum biomarker studies are noninvasive and can easily be used to measure changes in multiple biomarkers over time. However, the results may be confounded by the metabolism and excretion of tumor-derived biomarkers. Imaging is likely to complement these biomarker studies by allowing direct assessment of the tumor tissue without the interference of metabolic processes in plasma or normal organs.
Perhaps the most unique characteristic of PET-based biomarkers is that they measure a biological process and not the concentration of a particular cellular protein or of messenger RNA. Thus, PET is generally not a noninvasive form of immunohistochemistry. For some applications this may be seen as a disadvantage because the PET signal cannot invariably be attributed solely to the expression of a particular protein and may, therefore, appear to be unspecific. On the other hand, the ability to image a biochemical process in a living organism is probably the biggest strength of PET when compared to nonimaging biomarkers. Although continuous progress is being made, the ability to predict cellular function on the basis of gene or protein expression remains limited (2). Therefore, the best way for PET and other imaging biomarkers to complement gene and protein expression profiles may be to provide information about the functional consequences of certain alterations in these profiles and to monitor functional changes during therapeutic interventions. The other key advantages of PET include, when using fluorodeoxyglucose (FDG), it is quite reproducible and widely available, increasingly standard methods are being applied to assess the intrinsically quantitative images, and it can be used before treatment as well as during and after treatment. The ability to measure noninvasively the effects of a cancer therapy in an individual is a very attractive aspect of PET imaging.
Applications of PET as a Biomarker
The broad definition of biomarkers covers almost all applications of PET in oncology, including differentiation of benign and malignant tumors, tumor staging, and detection of recurrent disease. These aspects of PET imaging are covered in others chapters of this book for individual tumor types. In this chapter the focus is on the evaluation of tissue pharmacokinetics by radiolabeled drugs and assessment of tumor response to therapy.
Table 15.1 Overview of PET Imaging Agents that have been Used to Study Malignant Tumors in Patients | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Evaluation of the Tissue Pharmacokinetics
Labeling of drugs with positron-emitting radionuclides to study their pharmacokinetics was used in drug development even before PET became available. In 1973 Fowler et al. (3) described a procedure to radiolabel the then relatively new drug 5-fluorouracil (5-FU) with the positron emitter fluorine-18 ([18F]). In 1977 Shani and Wolf (4) demonstrated in a mouse model of leukemia that tumor uptake of 5-[18F]-fluorouracil ([18F]-5-FU) is significantly lower in resistant tumors than in sensitive tumors. They conclude that “non-invasive quantification of tumor/blood ratios following administration of [18F]5FU” may allow “differentiation of those human tumors that are likely to respond to drug therapy from those in which the response will be minimal or nil.”
Although this prediction has eventually been found to be correct, quantification of [18F]-5-FU has proven to be extremely complex. Rapid metabolism of injected [18F]-5-FU gives rise to high levels of labeled catabolites, which makes quantitative assessment of the [18F]-5-FU kinetics challenging. Thirty years after the publication of the study of radiosynthesis of [18F]-5-FU, the quantification of the tumor uptake and metabolism of [18F]-5-FU is still the subject of experimental studies.
Unfortunately, [18F]-5-FU is only one example of radiolabeled anticancer drugs with pharmacokinetics that are challenging to measure by PET imaging. Rapid metabolism and/or high unspecific accumulation are common problems when using radiolabeled anticancer drugs as imaging agents. Despite these difficulties, PET imaging with radiolabeled drugs has been successfully used to study tissue pharmacokinetics noninvasively in patients. For example, Saleem et al. (5) were able to quantify the increase of the intratumoral concentration of 5-FU in response to treatment with eniluracil, which inhibits the catabolism of 5-FU. Jayson et al. (6) used PET imaging to demonstrate the variability of tumor uptake of the iodine-124 labeled anti–vascular endothelial growth factor antibody HuMV833. PET has also been applied to study drug efflux by using various radiolabeled substrates of the mdr1 transporter. In animal models tumors with and without mdr1 expression could be differentiated by PET imaging. Furthermore, it was possible to monitor the inhibition of mdr1 function by specific antagonists. However, these encouraging findings still need to be confirmed in systematic clinical trials in patients with malignant tumors (7).
In summary, these studies demonstrate the feasibility of using PET with radiolabeled drugs for noninvasive pharmacokinetic studies. However, it is clear that radiolabeling strategies and techniques for PET image analysis need to be developed early in the course of drug development. Otherwise it is unlikely that a radiolabeled analogue of a drug will be available in time for the clinical testing of a new drug.
Monitoring Tumor Therapy
A significant number of studies have now shown that an abnormal FDG PET scan after completion of chemotherapy or chemoradiotherapy is associated with a high risk for early disease recurrence and poor prognosis. This has been demonstrated most extensively in patients with malignant lymphomas, but similar findings have been reported in a variety of solid tumors, such as cervical, esophageal, and lung cancers. In patients treated with neoadjuvant chemoradiotherapy, residual tumor FDG uptake has been shown to be correlated with the degree of histological tumor regression.
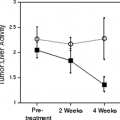
The observation that patients with a favorable response frequently show negative PET scans after completion of therapy, despite significant residual morphological abnormalities, has lead to the hypothesis that measurable quantitative changes in tumor metabolism may already occur during therapy, and that these may be used as an early indicator for tumor response. This hypothesis was initially evaluated by Wahl et al. (8) in patients with breast cancer undergoing neoadjuvant chemotherapy. The results of this study indicated that in responding tumors metabolic activity markedly changes within the first weeks of therapy. Subsequent studies by Jansson et al. (9) in breast cancer and by Findlay et al. (10) in colorectal cancer have also suggested that tumor glucose utilization is rapidly decreased by effective therapy. More recently the accuracy of FDG PET to predict response and patient survival has been evaluated in larger studies and other tumor types. The results of these trials are summarized in Table 15.2.
In vitro studies have suggested that chemotherapy and radiotherapy may cause a “metabolic flare phenomenon” (11,12). This phenomenon has been attributed to the activation of energy-dependent cellular repair mechanisms. On the basis of these data it has been recommended that assessment of tumor response should not be performed until several weeks after completion of therapy. In these in vitro studies, however, FDG uptake was measured per surviving cells, and in Higashi et al. (12) per surviving well and per tissue culture well (mimicking the whole tumor). This parallels the clinical situation, where the change in the PET signal is determined by a combination of decreased FDG uptake due to cancer cell death
plus potentially increased FDG uptake by surviving tumor cells. The study differs from the in vivo setting in that no inflammatory cells were present nor was delivery of the radiotracer an issue in the tissue culture wells. A metabolic flare phenomenon has been observed clinically in metastatic breast cancer treated with tamoxifen and was associated with a good response to therapy. This initial increase in tumor metabolic activity is likely due to the partial estrogenlike stimulatory activity of this antiestrogen, which is apparent during the initial days of treatment (13).
plus potentially increased FDG uptake by surviving tumor cells. The study differs from the in vivo setting in that no inflammatory cells were present nor was delivery of the radiotracer an issue in the tissue culture wells. A metabolic flare phenomenon has been observed clinically in metastatic breast cancer treated with tamoxifen and was associated with a good response to therapy. This initial increase in tumor metabolic activity is likely due to the partial estrogenlike stimulatory activity of this antiestrogen, which is apparent during the initial days of treatment (13).
Table 15.2 Prognostic Relevance of Quantitative Changes in Tumor FDG-Uptake During Chemo- or Chemoradiotherapy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
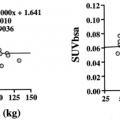
There are concerns that quantitative assessment of tumor glucose utilization by FDG PET is not reliable in clinical studies, since the measured signal is influenced by a variety of factors such as data acquisition and reconstruction protocols, lesion size, and blood glucose levels. However, for treatment monitoring it is only necessary to perform an intraindividual comparison of tumor FDG uptake. In this situation, many of the confounding factors will cancel out if the baseline and the follow-up scan are acquired and analyzed according to the same protocol. This is confirmed by studies indicating that the test/retest reproducibility of quantitative measurements of tumor FDG uptake in untreated patients is excellent, with a coefficient of variation of approximately 10% in the standard uptake values (SUV) (14,15).
FDG PET is also attractive for monitoring treatment with certain protein kinase inhibitors, since many signaling pathways targeted by protein kinase inhibitors also have a well-established role in regulating tumor glucose metabolism. For example the protein kinase Akt is a central regulator of cellular apoptosis (16,17), but it is also involved in the regulation of glucose utilization (18). Recent experimental data suggest that activation of Akt may be a key factor for the markedly increased glucose utilization of cancer cells (19,20).
FDG PET has already been used in clinical studies to monitor the response of gastrointestinal stromal tumors to treatment with imatinib (21,22,23,24). A marked reduction of tumor metabolic activity was noted as early as 24 hours after the first dose of imatinib (21,25). Moreover, extensive anatomical abnormalities observed by computed tomography (CT) persisted at a time when metabolic alterations had already been resolved. This rapid change in FDG uptake appears to be mediated by a translocation of glucose transporters from the plasma membrane to the cytosol and precedes cell death. Similar observations have been made in experimental models for epidermal growth factor receptor kinase inhibitors (26,27). These data suggest that FDG PET may become a valuable tool to monitor treatment with imatinib and potentially other protein kinase inhibitors.
Monitoring Tumor Cell Proliferation and Apoptosis
Despite the encouraging data, it is not clear whether there are similar marked changes for other forms of targeted therapy. For example, recent studies have indicated that antiangiogenic therapy may not significantly affect tumor glucose utilization, despite significant growth inhibition (28). There is considerable interest in techniques that more directly image proliferation and apoptosis, the major processes targeted by anticancer therapy.
The thymidine analogue [18F]-fluorothymidine (FLT) has been evaluated in several studies as a marker of tumor cell proliferation (29,30,31,32). Following cellular uptake FLT is phosphorylated by the cytosolic thymidine kinase 1 (TK1) and trapped intracellularly as FLT phosphate. In contrast to thymidine, FLT is not incorporated into the DNA. Nevertheless, FLT uptake is a marker of cellular proliferation, since significant TK1 activity is only observed during S phase.
Preclinical and clinical studies in untreated tumors have confirmed that tumor FLT uptake is generally well correlated with histologic markers of tumor cell proliferation. Animal studies have shown that inhibition of cellular proliferation by chemotherapy (33), radiotherapy (34,35), androgen deprivation (36), or ErbB1/2
kinase inhibition (37) leads to a rapid decrease of tumor FLT uptake that is correlated with ex vivo measurements of tumor cell proliferation. These data make FLT a promising imaging agent for monitoring the efficacy of cytostatic drugs. However, clinical experience with FLT PET for treatment monitoring is still limited. In patients, tumor FDG uptake is generally significantly lower than FLT uptake, which may make quantitative assessment of tumor cell proliferation challenging in tumors with relatively low baseline proliferative activity as the tumors cannot be clearly imaged distinct from surrounding normal tissue background levels.
kinase inhibition (37) leads to a rapid decrease of tumor FLT uptake that is correlated with ex vivo measurements of tumor cell proliferation. These data make FLT a promising imaging agent for monitoring the efficacy of cytostatic drugs. However, clinical experience with FLT PET for treatment monitoring is still limited. In patients, tumor FDG uptake is generally significantly lower than FLT uptake, which may make quantitative assessment of tumor cell proliferation challenging in tumors with relatively low baseline proliferative activity as the tumors cannot be clearly imaged distinct from surrounding normal tissue background levels.
Annexin V binds with high affinity to phosphatidyl serine, which is externalized during apoptosis. Single-photon emission computed tomography (SPECT) with technetium-99m-labeled annexin V has been used in small clinical studies to monitor tumor cell apoptosis induced by chemo- and radiotherapy. These studies have shown that the annexin V signal is frequently small and not easily detectable by SPECT imaging (38,39). This is not unexpected since apoptosis is a relatively rapid process. Therefore, even after effective therapy only a fraction of the tumor cells are expected to be in the apoptotic state. PET should provide higher sensitivity and resolution for imaging of annexin V binding than SPECT, and PET also provides the opportunity for quantitative assessment of annexin V uptake. Fluorine-18- or iodine-124-labeled annexin V analogues have been developed and validated in animal models (40,41). However, clinical studies using these PET-imaging agents have not yet been published. Since the time course of detectability of apoptotic cells may be brief, this appears to represent a challenging process to image.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


