References
Country
No. of patients
CCA site
Overall number of patients with P53 mutations (%)
Notes
Jonas et al. [23]
Germany
12
Perihilar
2 (16.6)
P53 exons 5–8 evaluated
Sturm et al. [22]
USA
27
Perihilar
7 (26.31)
P53 exons 5–8 evaluated
Petmitr et al. [21]
Thailand
20
IH-CCA
1 (5)
P53 exons 5–8 evaluated
Kang et al. [24]
Korea
40
IH-CCA
14 (35.7)
P53 exons 5–8 evaluated
Furubo et al. [25]
Japan
15
IH-CCA (peripheral) and perihilar
3a (20)
P53 exons 5–8 evaluated
Kamikawa et al. [19]
Japan
22
IH-CCA
9 (41.6)
P53 exons 5–8 evaluated; thorotrast exposed patients
Della Torre et al. [26]
Italy)
13
Not specified
2 (15.3)
P53 exons 5–8 evaluated
Tullo et al. [27]
Europe
29
Perihilar
7 (24)
P53 exons 5–8 evaluated
3/7 cases carried germline heterozygous polymorphism in tumoral and non-tumoral DNA
Momoi et al. [28]
Japan
28
IH-CCA
2 (7.1)
P53 exons 5–8 evaluated
Khan et al. [18]
UK
31
IH-CCA
24 (76)
Complete P53 mutational signatures
Three new frameshift mutations and two new intron mutations discovered
Liu et al. [29]
China
36
Not specified
22 (62)
P53 exons 5–8 evaluated
Kiba et al. [20]c
Thailand
26
IH-CCA
9 (35.7)
P53 exons 5–8 evaluated
2 patients with KRAS mutations, none carrying both P53 and KRAS mutations
Kiba et al. [20]c
Japan
12
IH-CCA
4 (33.3)
P53 exons 5–8 evaluated
7 patients with KRAS mutations, none carrying both P53 and KRAS mutations
Imai et al. [30]c
Japan
7
IH-CCAb
2 (28.5)
P53 exons 5–8 evaluated
Itoi et al. [31]c
Japan
12
Not specified
4 (33.3)
6 patients with KRAS mutations, none carrying both P53 and KRAS mutations
KRAS and P53 abnormalities not detected in non-neoplastic biliary tract tissues
The same mutation patterns detected in bile and neoplastic tissue
Total 330
Total 112 (34.0)
In the absence of definite environmental risk factors, P53 mutations are more frequent in areas with high CCA incidence (United States of America high-incidence cluster area = 67 %) than in areas with low incidence (United States low-incidence cluster = 20 %) [22]. This could reflect the exposure to unidentified mutagen triggering P53, in high-incidence areas. Unfortunately, very little is still known on environmental mutagens, and our current capability to disclose P53 impairment is limited. In the western world, similar rates (on average, 51 %) of P53 mutation have been found in CCA associated or not with PSC, indicating the lack of a PSC–CCA-specific molecular signature in P53 gene. It has been previously suggested that P53 alterations in CCA may be mediated by abnormal intracellular signaling cascades caused by cytotoxic biliary constituents [18]. In PSC, changes in bile composition are associated with bile duct inflammation and enhanced cholangiocyte proliferation, and this could favor, according to the clonal model of carcinogenesis, accumulation of mutations up to the threshold of neoplastic transformation. The alternative model of cholangiocarcinogenesis contemplates the involvement of individual genetic and environmental factors [17]. Several P53 polymorphisms have been so far described. Their relevance is unclear, and only two of these variants are associated with abnormal amino acid sequence of the P53 protein [18]. The lack of a specific P53 molecular signature in sporadic CCA could be explained if a definite gene polymorphism predisposes to P53 alterations in the presence of the pathological milieu (i.e., inflammation) determined by CCA risk factors. In comparison with the sporadic form, CCA associated with thorotrast exposure showed a different pattern of P53 mutations [18, 19]. It is, however, important to note that the full-length P53 cDNA has been insufficiently investigated. Indeed of the fourteen P53 sequencing studies, thirteen have evaluated only P53 exons 5–8, whereas the only study that evaluated the complete P53 mutational signatures disclosed three new frameshift mutations and two new intron mutations and demonstrated the highest mutation rate in P53 gene never reported (76 %).
In conclusion, the frequency and type of P53 mutations occurring in CCA patients depends from environmental factors, including the nature and dose of exposure to environmental carcinogens, which vary in different populations [18].
Growth factors and growth factor receptors (e.g., the ErbB family, insulin-like growth factors (IGF), and hepatocyte growth factor (HGF/MET)) are pivotal growth signal regulators in cancers of different origin [10]. Among the pathways involved in the pathogenesis of IH-CCA, the family of ErbB receptors is perhaps the most relevant [10, 11]. ErbB-2 is an epidermal growth factor receptor (EGFR) homologue and is able to homodimerize or heterodimerize with other members of the EGFR superfamily, resulting in activation of the Raf/MAPK pathway [10, 11]. The most notable are the aberrant regulation of ErbB2 and the EGFR signaling [10, 11]. Constitutive overexpression of ErbB2 and/or ErbB1 in malignant cholangiocytes has been documented in more than 50 % of IH-CCA [32, 33]. In addition, experimental models of IH-CCA in rodents are associated with constitutive ErbB2 overexpression [11]. ErbB2 and ErbB1 interact with different relevant molecular signaling pathways associated with IH-CCA development and progression, including bile acids, IL (interleukin)-6/gp130, transmembrane mucins, HGF/MET, and vascular endothelial growth factor (VEGF) signaling [11, 32, 33]. Hydrophobic bile salts, such as deoxycholate, may play a carcinogenetic role through transactivation of EGFR and impairment of Mcl-1 functions, and this has been considered a mechanism favouring the intraductal pattern of growth characterizing a subset of CCAs [11]. The relevance of ErbB2- or ErbB1-related pathways in CCA has raised interest in exploring, for the treatment of CCA, agents selectively targeting these receptors. However, current experience with ErbB-targeted therapies produced only modest responses in patients with biliary tract cancers [10, 11]. Activation of EGFR triggers downstream Ras/Raf/Mek/Erk and PI3K/PTEN/Akt, two major cell survival pathways. Ras proteins (K-Ras, N-Ras, H-Ras, B-Raf), responsible for signal transduction downstream to growth factor receptors, have been largely investigated in CCA, and in this regard, KRAS-activating mutations represent one of the most frequent genetic alterations found in CCA (10–75 % of CCA cases) [34]. After binding and activation by GTP, Ras proteins recruit Raf that, in turn, activates, by phosphorylation, MAP kinases (MEK1/2 and ERK1/2) [10, 11]. Activation of MAP kinase pathways leads to enhanced proliferation and inhibition of apoptosis.
As reported in Table 2, a total number of 218 CCA patients have been investigated by sequencing studies aimed to identify KRAS mutations [20, 30, 31, 35–39, 40, 41]. Studies from 1992 to 2011 have evaluated CCA patient courts from Europe, America, and Asia, as shown in Table 2 [20, 30, 31, 35–39, 40, 41]. The total number of CCA patients with KRAS mutations resulted 88, the 40.4 % of all the CCAs. When classified by tumor site, 17 % of peripheral type CCAs were positive for KRAS mutations with the most frequent alteration in codon 12. Importantly, the incidence of mutations was higher in the hilar-type tumors (53 %) [34]. It is noteworthy that the frequency of KRAS mutations increases with tumor stage (stage I, 8 %; stage II, 15 %; stage III, 31 %; stage IV, 46 %) [39].
Table 2
KRAS mutations in human cholangiocarcinoma: sequencing studies
References | Country | No. of patients | CCA site | Overall number of patients with KRAS mutations (%) | Notes |
|---|---|---|---|---|---|
Tada et al. [35] | Japan | 18 | IH-CCA (peripheral) and perihilar | 9 (50) | The incidence of mutations higher in the perihilar CCA |
Tannapfel et al. [37] | Germany | 41 | IH-CCA | 22 (54) | All 22 cancers with KRAS mutations also exhibited methylated P16; in 2 cases, mutations were detected in non-neoplastic liver tissue surrounding the tumor (germline mutations) |
Ahrendt et al. [38] | USA | 12 | Not specified | 12 (33) | Patients with PSC-associated CCA |
Overall survival shorter in patients with KRAS mutation | |||||
Xu et al. [39] | China | 13 | Not specified | 5 (38.2) | 2 patients (5.9 %) harbored both KRAS and PIK3CA mutations |
Isa et al. [41] | Japan | 23 | IH-CCA (peripheral) and perihilar | 9 (39.1) | Patients with KRAS mutations worst survival rates; KRAS mutation rates higher in perihilar (6/8, 75.0 %) than in peripheral (3/5, 20.0 %) CCA |
Rashid et al. [40] | China | 33 | Not specified | 5 (15.2) | Mean survival of patients with KRAS mutations shorter (3.0 months) compared with patients without mutation (15.5 months) |
Kiba et al. [20]b | Thailand | 26 | IH-CCA | 2 (7.6) | P53 exons 5–8 also evaluated; 9 patients (35.7 %) with P53 mutations |
Kiba et al. [20]b | Japan | 12 | IH-CCA | 7 (58.4) | P53 exons 5–8 also evaluated and the overall number of patients with P53 mutations was 4 (33.3 %) |
Ohashi et al. [36]b | Japan | 21 | IH-CCA | 10 (48) | P53 exons 5–8 also evaluated; 2 patients (7.1 %) with P53 mutations; KRAS mutations were prominent in the periductal growing CCA (4/6; 67 %) with respect to the mass-forming CCA (0/5) |
Imai et al. [30]b | Japan | 7 | IH-CCAa | 1 (14.2) | P53 exons 5–8 also evaluated; 2 patients (28.5 %) with P53 mutations |
Itoi et al. [31]b | Japan | 12 | Not specified | 6 (50) | P53 exons 5–8 also evaluated; 4 patients (33.3 %) with P53 mutations |
KRAS abnormalities were not detected in non-neoplastic tissues | |||||
The same mutation patterns detected in bile and neoplastic tissues | |||||
Total 218 | 88 (40.4) |
Another recently proposed mechanism linking chronic inflammation with CCA development is related to activation-induced cytidine deaminase (AID), a member of the DNA/RNA editing enzyme family, implicated in human cancerogenesis via its mutagenic activity [42]. AID was found to be increased in biopsies from patients with PSC or CCA, whereas only trace amounts of AID were detected in the normal liver [11, 42]. In in vitro studies, in human CCA cell lines, AID was induced by tumor necrosis factor-alpha that, in turn, was stimulated via IkappaB kinase-dependent nuclear factor-kappaB (NF-kappaB) pathway [11]. The aberrant expression of AID in biliary cells resulted in the generation of somatic mutations in tumor-related genes, including P53, c-Myc, and the promoter region of the INK4A/P16 sequences [10, 11]. In contrast with hepatocellular carcinoma (HCC), mutations activating 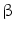 -catenin are rarely found in CCA (0–8 % of CCA cases) [10]. Other genes such as IDH1, SMAD4, and KEAP1 have been described to be frequently mutated in CCA tissue, but with large differences among studies. [10, 11, 43]. Aberrant epigenetic regulation, such as promoter hypermethylation, was demonstrated in numerous important cancer-associated genes in CCA [44, 45]. Promoter methylation of P14, a regulator of P53, has been found in CCA [10]. P16 (CDKN2) is frequently silenced in CCA by genetic or epigenetic mechanisms [37].
-catenin are rarely found in CCA (0–8 % of CCA cases) [10]. Other genes such as IDH1, SMAD4, and KEAP1 have been described to be frequently mutated in CCA tissue, but with large differences among studies. [10, 11, 43]. Aberrant epigenetic regulation, such as promoter hypermethylation, was demonstrated in numerous important cancer-associated genes in CCA [44, 45]. Promoter methylation of P14, a regulator of P53, has been found in CCA [10]. P16 (CDKN2) is frequently silenced in CCA by genetic or epigenetic mechanisms [37].
-catenin are rarely found in CCA (0–8 % of CCA cases) [10]. Other genes such as IDH1, SMAD4, and KEAP1 have been described to be frequently mutated in CCA tissue, but with large differences among studies. [10, 11, 43]. Aberrant epigenetic regulation, such as promoter hypermethylation, was demonstrated in numerous important cancer-associated genes in CCA [44, 45]. Promoter methylation of P14, a regulator of P53, has been found in CCA [10]. P16 (CDKN2) is frequently silenced in CCA by genetic or epigenetic mechanisms [37].The interleukin-6 (IL-6) is one of the most investigated genes in the pathogenesis of CCA, where it could be involved by different mechanisms [10, 11]. IL-6 is produced at high levels in CCA cells and elevated IL-6 serum concentrations have been reported in CCA patients [10, 11]. Constitutive activation of the IL-6/STAT3 pathway has been described in CCA cells, and this was associated with silencing of SOCS3. The methylation of SOCS3 promoters occurs in 61 % of IH-CCA together with down-regulation of gp130, a membrane protein that, when associated with SOCS3 protein product, inhibits the IL-6 pathway [44]. By autocrine and paracrine mechanisms, IL-6 activates via STAT3 the prosurvival P38 mitogen-activated protein kinase [10, 11]. STAT3 is an activator of p44/42 and P38 mitogen-activated protein kinase that has been frequently found, by immunohistochemistry, to be activated in IH-CCA [10, 11]. In addition, IL-6 up-regulated the expression of myeloid cell leukemia-1 (Mcl-1) through STAT3- and AKT-related signaling pathways [46, 47]. Mcl-1 increases cell resistance to TRAIL apoptotic signals [48]. Moreover, IL-6-related pathways can modulate epigenetic fate of the cells through DNA (cytosine-5)-methyltransferase 1 (DNMT1), and this has been demonstrated for IL-6-mediated up-regulation of EGFR and for down-regulation of P53 expression, which occur by promoter hypo- or hypermethylation, respectively [10, 12]. Finally, IL-6 may act in CCA by autocrine and paracrine pathways since it is secreted by malignant cholangiocytes [11]. In light of these findings, IL-6 has been explored in the diagnostic setting and, in fact, serum levels of IL-6 have been correlated with tumor burden in CCA patients [13]. However, although these findings are encouraging, it should be considered that serum IL-6 is also elevated in many patients with HCC, benign biliary disease, and metastatic lesions, and therefore, the specificity of high IL-6 serum levels for CCA is still debated [13]. Recently, the induction of progranulin (PGRN) has been advanced as another mechanism by which IL-6 could enter CCA pathogenesis [49]. PGRN is involved in multiple steps of the tumor progression cascade, including cellular proliferation, anchorage independence, invasiveness, resistance to apoptosis, and promotion of resistance to certain cytotoxic drugs. In addition, PGRN may also act by promoting neoangiogenesis with a direct effect on endothelial cells as well as an indirect effect on VEGF synthesis. The expression and secretion of PGRN are up-regulated in human CCA, and this in part occurs via IL-6-mediated activation of the Erk1/2/Rsk1/C/EBPb pathway [49]. Serum PGRN levels were higher in patients with CCA than in non-neoplastic controls, but it is unknown if this can discriminate CCA with respect to benign biliary pathologies, including PSC and benign strictures of the biliary tree [13]. IL-6 and other mediators of inflammation, including TNF-alpha, may enter CCA pathogenesis by inducing or synergizing a number of different growth factors [10, 11].
Cyclooxygenase 2 (COX-2), the rate-limiting enzyme in prostaglandin biosynthesis from arachidonic acid, activated by inflammatory cytokines and nitric oxide (NO), accelerates cell cycle via prostaglandin E2 (PGE2) and inhibits different apoptotic cascades. Indeed, increased COX-2 immunohistochemical expression has been documented in more than 70 % of CCA samples [50], and the COX-2 gene is frequently affected by epigenetic (methylation) perturbations in CCA. COX-2 is activated by oxysterols, oxygenated cholesterol derivatives formed in the bile of patients with inflammatory diseases of the biliary tree, and by hydrophobic bile acids [11]. Another COX-2-inducing molecule is the tyrosine kinase ErbB-2, which is overexpressed in CCA and involved in CCA origin and progression [11]. Current evidence supports a primary role played by NO, induced by proinflammatory cytokines (TNF-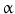 , IL-6, etc.) [51]. These cytokines are able to activate inducible nitric oxide synthase (iNOS), which, at the immunohistochemical level, is overexpressed in more than 70 % CCA [11]. Increased iNOS activity results in generations of NO and reactive oxygen species, which are known to interact with cellular DNA and to inhibit DNA reparative mechanisms, thus triggering oncogenetic mutations. NO together with different cytokines can also inhibit cholangiocyte apoptosis by nitrosylation of caspase-9 and may also induce proliferation, thus favouring accumulation of somatic mutations [11]. Very recently, a relevant role in modulating CCA growth and proliferation has been attributed to estrogens, IGF1, leptin, opioid receptor modulators, endothelin, and serotonin [11]. As far as estrogens are concerned, recent studies suggest their synergistic action with growth factors (IGF1, VEGF) in sustaining the cholangiocyte proliferative machinery and in depressing apoptosis [52, 53]. Indeed, a cross talk between IGF1 and estrogens has been demonstrated to modulate CCA proliferation, whereas estrogens act at several points of the IGF1 signal transduction pathway [52]. In addition, it has been shown that the estrogen proliferative effect on CCA cells is also due to the stimulation of VEGF synthesis and secretion [52, 53]. In agreement with these data, IGF1 have been explored as CCA markers in a diagnostic setting. The IGF1 biliary concentration was shown to be capable of completely discriminating CCA from benign biliary pathologies and pancreatic cancer [54].
, IL-6, etc.) [51]. These cytokines are able to activate inducible nitric oxide synthase (iNOS), which, at the immunohistochemical level, is overexpressed in more than 70 % CCA [11]. Increased iNOS activity results in generations of NO and reactive oxygen species, which are known to interact with cellular DNA and to inhibit DNA reparative mechanisms, thus triggering oncogenetic mutations. NO together with different cytokines can also inhibit cholangiocyte apoptosis by nitrosylation of caspase-9 and may also induce proliferation, thus favouring accumulation of somatic mutations [11]. Very recently, a relevant role in modulating CCA growth and proliferation has been attributed to estrogens, IGF1, leptin, opioid receptor modulators, endothelin, and serotonin [11]. As far as estrogens are concerned, recent studies suggest their synergistic action with growth factors (IGF1, VEGF) in sustaining the cholangiocyte proliferative machinery and in depressing apoptosis [52, 53]. Indeed, a cross talk between IGF1 and estrogens has been demonstrated to modulate CCA proliferation, whereas estrogens act at several points of the IGF1 signal transduction pathway [52]. In addition, it has been shown that the estrogen proliferative effect on CCA cells is also due to the stimulation of VEGF synthesis and secretion [52, 53]. In agreement with these data, IGF1 have been explored as CCA markers in a diagnostic setting. The IGF1 biliary concentration was shown to be capable of completely discriminating CCA from benign biliary pathologies and pancreatic cancer [54].
, IL-6, etc.) [51]. These cytokines are able to activate inducible nitric oxide synthase (iNOS), which, at the immunohistochemical level, is overexpressed in more than 70 % CCA [11]. Increased iNOS activity results in generations of NO and reactive oxygen species, which are known to interact with cellular DNA and to inhibit DNA reparative mechanisms, thus triggering oncogenetic mutations. NO together with different cytokines can also inhibit cholangiocyte apoptosis by nitrosylation of caspase-9 and may also induce proliferation, thus favouring accumulation of somatic mutations [11]. Very recently, a relevant role in modulating CCA growth and proliferation has been attributed to estrogens, IGF1, leptin, opioid receptor modulators, endothelin, and serotonin [11]. As far as estrogens are concerned, recent studies suggest their synergistic action with growth factors (IGF1, VEGF) in sustaining the cholangiocyte proliferative machinery and in depressing apoptosis [52, 53]. Indeed, a cross talk between IGF1 and estrogens has been demonstrated to modulate CCA proliferation, whereas estrogens act at several points of the IGF1 signal transduction pathway [52]. In addition, it has been shown that the estrogen proliferative effect on CCA cells is also due to the stimulation of VEGF synthesis and secretion [52, 53]. In agreement with these data, IGF1 have been explored as CCA markers in a diagnostic setting. The IGF1 biliary concentration was shown to be capable of completely discriminating CCA from benign biliary pathologies and pancreatic cancer [54].Recent technical improvement in molecular profiling platforms is adding new insights into the current knowledge of cholangiocarcinogenesis favoring the integration of the different proposed models. Unfortunately, few comparative genomic hybridization (CGH) studies on CCA have been performed during the past decade, and these studies are biased by the heterogeneous population investigated that included IH-CCA, EH-CCA, or even gallbladder cancers, making difficult any accurate interpretation. Evaluation of DNA copy number (CN) demonstrated CN gains in the region of several molecular targets: ERBB2, MEK2, PDGFB, MTOR, VEGFR-3, PDGFA, RAF1, VEGFA, and EGFR [55]. Technological advances also allow the differential characterization of genomic and genetic features of CCA epithelial and stromal compartments [56]. The tumor epithelium was defined by deregulation of the HER2 network and frequent overexpression of EGFR, the HGF/MET receptor, pRPS6, and Ki67, whereas stroma was enriched in inflammatory cytokines [56]. Recently, the comparative evaluation of gene expression profile (transcriptome), clinicopathological traits, and patient outcomes in IH-CCA cases has allowed the identification of 2 main biologic classes of IH-CCA: (1) the inflammation class (38 % of IH-CCA), characterized by activation of inflammatory signaling pathways, overexpression of cytokines, and STAT3 activation and (2) the proliferation class (62 % of IH-CCA), characterized by activation of oncogenic signaling pathways (i.e., RAS, MAP kinase, and HGF/MET), DNA amplifications at 11q13.2, deletions at 14q22.1, mutations in KRAS and BRAF, and gene expression signatures previously associated with poor outcomes for patients with HCC [57]. As previously discussed, an optimal approach to CCA molecular profiling should be the comparative investigation of subtypes such as CCA emerging in a definite category at risk, including PSC or liver fluke infestation. Unfortunately, very few studies followed this type of approach. PSC is a major risk factor for IH- and EH-CCAs, and these patients experienced a cumulative risk of 11.2 %, 10 years after diagnosis [7]. Unfortunately, predictive factors or standardized screening or surveillance strategies are lacking. Different molecular signatures of the high oncogenic risk have been described in PSC patients. KRAS mutations have been found in 30 % of bile fluid of PSC patients without evidence of CCA [58]. Since KRAS mutations are frequently observed in CCA, this could be an early event of bile duct carcinogenesis in PCS patients. Notably, mutational profiling can be performed in cell-free DNA of bile supernatant [59]. The inflammatory microenvironment has also been associated with an aberrant DNA methylation profile in PSC-derived CCA, which provides survival signals for the tumor [60]. Genetic susceptibility of PSC patients for CCA development has been demonstrated by studies concerning the natural killer cell receptor G2D receptor [61], where specific genetic variants have been described in PSC patients.
The association between liver flukes and CCA has been evaluated by the International Agency for Research on Cancer (IARC) since 1994. Opisthorchis viverrini (OV) infestation, endemic in Southeast Asia, is now considered a definitive carcinogen. The molecular mechanism of OV-associated CCA has been also studied in experimental models. Up-regulation of 23 transcripts and down-regulation of 1 transcript related to CCA induced in OV-infected hamsters has been identified. The up-regulated genes include signal transduction protein kinase A regulatory subunit Ia (PRKAR1a), myristoylated alanine-rich protein kinase C substrate, transcriptional factor LIM-4-only domain, oxysterol-binding protein involved in lipid metabolism, splicing regulatory protein 9, ubiquitin-conjugating enzyme involved in protein degradation, 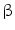 -tubulin,
-tubulin, 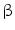 -actin, and collagen type VI. Interestingly, PRKAR1a expression tended to increase during the progression from hyperplasia to precancerous lesions and to CCA [62]. In humans, molecular studies of IH-CCA associated with liver flukes demonstrated overexpression of genes involved in xenobiotic metabolism (UGT2B11, UGT1A10, CHST4, SULT1C1), whereas, in contrast, non-OV-associated IH-CCA showed enhanced expression of genes related to growth factor signaling (TGFBI, PGF, IGFBP1, IGFBP3). Thus, the evaluation of the putative signature of OV-associated IH-CCA in OV-infected patients could help in screening and surveillance, with the perspective of an early diagnosis [63]. The draft genome of Clonorchis sinensis and transcriptomes of Clonorchis sinensis and OV have been recently elucidated [64, 65]. Recently, a study in a large IH-CCA cohort (N = 102) associated with liver fluke infection demonstrated promoter hypermethylation in a handful of target genes, when CCA specimens were compared with adjacent non-tumoral tissues [66]. These results could help in identifying molecules linked with the development of liver fluke-induced CCA. CCA genetic susceptibility has been investigated in geographic areas endemic for liver flukes. In these studies, specific haplotypes of COX-2-coding gene (PTGS2) or IL8RB have been recently associated with a significant risk of CCA development [67].
-actin, and collagen type VI. Interestingly, PRKAR1a expression tended to increase during the progression from hyperplasia to precancerous lesions and to CCA [62]. In humans, molecular studies of IH-CCA associated with liver flukes demonstrated overexpression of genes involved in xenobiotic metabolism (UGT2B11, UGT1A10, CHST4, SULT1C1), whereas, in contrast, non-OV-associated IH-CCA showed enhanced expression of genes related to growth factor signaling (TGFBI, PGF, IGFBP1, IGFBP3). Thus, the evaluation of the putative signature of OV-associated IH-CCA in OV-infected patients could help in screening and surveillance, with the perspective of an early diagnosis [63]. The draft genome of Clonorchis sinensis and transcriptomes of Clonorchis sinensis and OV have been recently elucidated [64, 65]. Recently, a study in a large IH-CCA cohort (N = 102) associated with liver fluke infection demonstrated promoter hypermethylation in a handful of target genes, when CCA specimens were compared with adjacent non-tumoral tissues [66]. These results could help in identifying molecules linked with the development of liver fluke-induced CCA. CCA genetic susceptibility has been investigated in geographic areas endemic for liver flukes. In these studies, specific haplotypes of COX-2-coding gene (PTGS2) or IL8RB have been recently associated with a significant risk of CCA development [67].
-tubulin, -actin, and collagen type VI. Interestingly, PRKAR1a expression tended to increase during the progression from hyperplasia to precancerous lesions and to CCA [62]. In humans, molecular studies of IH-CCA associated with liver flukes demonstrated overexpression of genes involved in xenobiotic metabolism (UGT2B11, UGT1A10, CHST4, SULT1C1), whereas, in contrast, non-OV-associated IH-CCA showed enhanced expression of genes related to growth factor signaling (TGFBI, PGF, IGFBP1, IGFBP3). Thus, the evaluation of the putative signature of OV-associated IH-CCA in OV-infected patients could help in screening and surveillance, with the perspective of an early diagnosis [63]. The draft genome of Clonorchis sinensis and transcriptomes of Clonorchis sinensis and OV have been recently elucidated [64, 65]. Recently, a study in a large IH-CCA cohort (N = 102) associated with liver fluke infection demonstrated promoter hypermethylation in a handful of target genes, when CCA specimens were compared with adjacent non-tumoral tissues [66]. These results could help in identifying molecules linked with the development of liver fluke-induced CCA. CCA genetic susceptibility has been investigated in geographic areas endemic for liver flukes. In these studies, specific haplotypes of COX-2-coding gene (PTGS2) or IL8RB have been recently associated with a significant risk of CCA development [67].3 Molecular Profiling and the Diagnosis of Cholangiocarcinoma
Immunohistochemical markers specific to CCA are lacking, and the definite diagnosis in bioptic or surgical samples is still based on a panel of markers aimed at excluding HCC or metastatic cancer. Therefore, for many years, studies have been focused on the search for CCA-specific markers. Different proposals appear in recent literature, but none of these reached clinical routine application. Recently, high-throughput techniques based on DNA microarray technology [68] have been tested in human CCA samples. The first study using DNA microarray technology (Affymetrix U133A) in a series of surgically resected biliary cancers, biliary cancer cell lines, and biliary epithelial scrapings was carried out in 2003 by Hansel et al. [69]. They reported 282 genes overexpressed threefold or greater in biliary malignancies or cancer cell lines, including proliferation and cell cycle-related genes (e.g., cyclins D2 and E2, cdc2/p34, and geminin genes), transcription factors (e.g., homeobox B7 and islet-1), growth factors and growth factor receptors (e.g., hepatocyte growth factor, amphiregulin, and insulin-like growth factor 1 receptor), two important downstream mediators of the mitogenic Akt/mTOR signaling pathway (ribosomal protein S6 kinase and eukaryotic translation initiation factor 4E), enzymes modulating sensitivity to chemotherapeutic agents (e.g., cystathionine beta synthase, dCMP deaminase, and CTP synthase), and cytosolic phospholipase A2 [69]. After this first report, other studies aimed to investigate the utility of transcriptomic in CCA diagnosis have been performed. A genome-wide cDNA microarray containing 27,648 cDNAs carried out in IH-CCA specimens and non-cancerous biliary tissues, showed 52 genes up-regulated and 421 genes down-regulated. The overexpressed genes are related to a variety of functions, such as signal transduction (GNAZ, MDK), transcription (FOXM1, HOXB7, DRIL1), DNA synthesis (TOP2A, TOP2B, NAV2, BUB1B, CKS2), antiapoptosis (BIRC5, S100P), angiogenesis (ECGF1), cytoskeleton (FSCN1, PRC1, ANLN, KIF2C), and cytokinesis or adhesion (CDH3, CIT, ECT2). On the contrary, the down-regulated genes are mainly involved in growth suppression (EGR1 and EGR2, AXIN1, AXUD1, DLC1, DOC1). From the 52 up-regulated genes, P-cadherin and survivin were selected for further investigation, and the enhanced expression of their protein products in CCA tissues was demonstrated by immunohistochemical staining [70]. Recently, oligonucleotide arrays (Affymetrix U133A) were used to establish a specific gene expression profile of IH-CCA in comparison with adjacent non-malignant liver tissue. Most of the strongly overexpressed genes are related to cell cycle regulation and DNA replication (15 genes, including ribonucleosidediphosphate reductase M2, calgizzarin, calcyclin, BUB1B) intracellular signaling (15 genes, including CD24 and MARCKS), genes encoding transcription factors (6 genes, such as SOX9), or genes involved in nuclear organization and nucleic metabolism (13 genes, such as thymidylate synthetase). Other up-regulated genes include those coding for extracellular matrix and cell adhesion molecules (37 genes, for example OPN, ADAM9, thymosin beta–10, integrin alpha–6), cytoskeleton structure proteins (16 genes, such as tropomyosin 2, cytokeratin 7 and 19), or enzymes involved in protein biosynthesis (4 genes). The gene encoding for OPN was identified as the highest and most consistently overexpressed gene (33.5-fold change) in all analyzed CCA samples. Most of the genes encoding proteins involved in cellular apoptosis (7 genes including growth arrest-specific protein 2, CIDE–B) were found to be down-regulated in IH-CCA [71]. The genes overexpressed in IH-CCA, have been confirmed at protein level by immunohistochemical analysis, and included osteopontin, P38 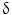 /MAPK-13, cadherin, and survivin. In conclusion, oligonucleotide microarray analysis shows a specific gene expression profile of IH-CCA, which could discriminate this cancer with respect to other malignancies or non-malignant lesions. These data, however, need further validation in independent cohorts of samples.
/MAPK-13, cadherin, and survivin. In conclusion, oligonucleotide microarray analysis shows a specific gene expression profile of IH-CCA, which could discriminate this cancer with respect to other malignancies or non-malignant lesions. These data, however, need further validation in independent cohorts of samples.
/MAPK-13, cadherin, and survivin. In conclusion, oligonucleotide microarray analysis shows a specific gene expression profile of IH-CCA, which could discriminate this cancer with respect to other malignancies or non-malignant lesions. These data, however, need further validation in independent cohorts of samples.The differential diagnosis between IH-CCA and some subtypes of HCC is frequently challenging because of the existence of many overlapping features. Indeed, detailed studies on immunohistochemical profile have revealed that a whole range of phenotypical traits of hepatocytes, cholangiocytes, and progenitor cells can be shared by IH-CCA, combined HCC-CCA, fibrolamellar HCC, and HCC with stem cell features. This is consistent with a common origin of these cancers from the hepatic stem cell compartment within canals of Hering [72]. A substantial number of HCCs, ranging from 28 to 50 % of human HCCs, express markers of progenitor cells or cholangiocytes including CK7, CK19, and OV6, which suggest an origin from bipotential stem/progenitor cells located within canals of Hering [73]. Some of these markers in HCC, especially CK19, have been associated with a worse prognosis and higher rates of recurrence after surgical treatment [73]. The emergence of HCC and IH-CCA in the same pathological context of chronic liver diseases does not help in differential diagnosis, and radiologic features may overlap. Differential diagnosis between HCC and IH-CCA deserves important clinical implications since, for example, IH-CCA is excluded from liver transplantation programs. Recently, mutations of BRAF and KRAS were evaluated in 25 HCC and in 69 CCA by direct DNA sequencing analyses after microdissection. Using this molecular profiling approach, RAS or BRAF mutations have been detected in approximately 62 % of CCA, but not in HCC [74]. The diagnostic utility of evaluation of active intermediates of the MAPK pathway was assessed by microarray gene expression. The study identified a P38 MAP kinase, P38 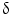 (also known as MAPK13 or SAPK4) as a protein that is up-regulated in CCA relative to HCC and to normal biliary tract tissues. Consistently, P38
(also known as MAPK13 or SAPK4) as a protein that is up-regulated in CCA relative to HCC and to normal biliary tract tissues. Consistently, P38 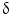 immunohistochemical staining distinguished CCA from HCC with a sensitivity of 92.6 % and a specificity of 90.7 %. P38
immunohistochemical staining distinguished CCA from HCC with a sensitivity of 92.6 % and a specificity of 90.7 %. P38 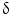 is important for motility and invasion of CCA cells, suggesting an important role in CCA metastasis. Therefore, P38
is important for motility and invasion of CCA cells, suggesting an important role in CCA metastasis. Therefore, P38 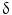 could represent a novel diagnostic marker for CCA and may also serve as a new target for molecular-based targeted therapy [75]. Evaluation of markers of apoptosis and cell proliferation, such as bcl-2, c-myc, Fas, Lewis(y), and P53
could represent a novel diagnostic marker for CCA and may also serve as a new target for molecular-based targeted therapy [75]. Evaluation of markers of apoptosis and cell proliferation, such as bcl-2, c-myc, Fas, Lewis(y), and P53




(also known as MAPK13 or SAPK4) as a protein that is up-regulated in CCA relative to HCC and to normal biliary tract tissues. Consistently, P38 immunohistochemical staining distinguished CCA from HCC with a sensitivity of 92.6 % and a specificity of 90.7 %. P38 is important for motility and invasion of CCA cells, suggesting an important role in CCA metastasis. Therefore, P38 could represent a novel diagnostic marker for CCA and may also serve as a new target for molecular-based targeted therapy [75]. Evaluation of markers of apoptosis and cell proliferation, such as bcl-2, c-myc, Fas, Lewis(y), and P53
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
