Progress in Amyloid Imaging
Brian J. Lopresti
William E. Klunk
Chester A. Mathis
Alzheimer’s disease (AD) is the most prevalent form of dementia afflicting the elderly population and is characterized by a progressive loss of memory and cognitive function. AD is one of the leading causes of death in industrialized societies, and afflicts more than 4 million Americans and an estimated 30 million people worldwide (1,2,3,4). Presently, a definitive diagnosis of AD can only be made at autopsy when the presence of the pathologic hallmarks of the disease, amyloid plaques, and neurofibrillary tangles (NFTs) can be confirmed (5).
Brief Survey of Alzheimer’s Disease Pathology
Amyloid plaques are comprised of fibrillar deposits of the amyloid-β (Aβ) peptide, which varies from 40-42 amino acids in length (6). Amyloid plaques can be classified as either diffuse or neuritic, the latter being characterized by a dense central core of Aβ fibrils that stain with dyes such as Congo red or thioflavin-S and are surrounded by glial cells and abnormal neuritic processes. NFTs are comprised of intraneuronal fibrillar deposits of a hyperphosphorylated form of the microtubule-associated protein tau (7), which have a characteristic looplike appearance and are also stained with Congo red and thioflavin-S.
Plaques occur earliest in the neocortex, where they are relatively evenly distributed (8), while NFTs appear first in limbic areas, such as the transentorhinal cortex and progress in a predictable pattern to the neocortex (9). Topological studies of the distribution of neuritic plaques and NFTs in AD brains revealed plaques to be more evenly distributed throughout the cortex than NFTs. The exceptions are the limbic periallocortex and allocortex (including the hippocampal formation), where NFTs are most concentrated and neuritic plaques are much less abundant (10,11). The cerebellum was found to be free of neuritic plaques and NFTs, even in late-stage AD, although it is not uncommon for diffuse cerebellar amyloid deposits to be observed in cerebellum (12,13).
The vast majority of cases of AD can be classified as sporadic, as no clear genetic or environmental causes have been identified. For sporadic AD, the most significant risk factor for developing the disease is age, with prevalence increasing from approximately 5% at age 70 to as high as 45% at age 85 (14). A small fraction of cases of AD (approximately 1%) can be attributed to specific genetic mutations with autosomal-dominant inheritance (15,16). These mutations occur in domains that encode the amyloid precursor protein (APP), the precursor to the 40 to 42 amino acid Aβ peptides that comprise amyloid plaques, or in domains encoding the presenilin proteins PS-1 and PS-2 that are believed to be involved in APP processing. These rare cases, classified as early onset familial AD (eoFAD), result in early AD pathology with 100% penetrance in gene carriers.
Although eoFAD does not represent a dominant public health concern, the study of this small population is particularly interesting as it permits the identification of a pool of subjects (carriers of the mutations) who are ensured of developing AD pathology years or decades before the onset of symptoms. Longitudinal assessments of brain amyloid burden using a noninvasive imaging agent in eoFAD subjects may provide a unique opportunity to elucidate the natural history of brain amyloid deposition.
Mild cognitive impairment (MCI) is a condition characterized by isolated memory impairment or impairments in several cognitive domains. The cognitive impairments are similar to those seen in patients with AD, but they are not severe or pervasive enough to meet the diagnostic criteria for AD (17). Although MCI patients convert to AD at a rate of 10% to 15% per year, 30% to 40% of MCI patients remain stable or do not convert to AD over 5 to 10 years (18). A great deal of effort has been directed toward detecting what clinical characteristics of MCI are predictive of conversion to AD,
but a variety of underlying pathologic substrates and the relative mildness and heterogeneity of clinical symptoms have complicated these efforts.
but a variety of underlying pathologic substrates and the relative mildness and heterogeneity of clinical symptoms have complicated these efforts.
Role of Amyloid in Alzheimer’s Disease Target for Therapeutics
Aβ plaques in the brain are a pathologic hallmark of the dementia first characterized by Alois Alzheimer’s in 1906, and a considerable body of evidence implicates elevated Aβ levels and the deposition of Aβ plaques as central events in the pathogenesis of AD (19). Perhaps the most compelling piece of evidence supporting this “amyloid cascade hypothesis” is that rare mutations in chromosome 21 result in abnormal processing of APP and cause eoFAD (20). Also compelling is the observation that mutations in the region of chromosome 14 that codes PS-1, which is believed to be an essential component of the γ-secretase complex that cleaves Aβ from APP, are the most common genetic causes of autosomal dominant eoFAD (21). Thus, these rare eoFAD cases have established that altered metabolism of APP results in increased Aβ and clinical AD.
A logical corollary of the amyloid cascade hypothesis is that decreased cerebral levels of Aβ should help prevent or delay the onset of AD. Indeed, the development of antiamyloid therapies is an active area of drug discovery and investigation. Some prospective antiamyloid therapies focus on means to decrease production of Aβ by inhibiting the β- or γ-secretase enzymes, which are responsible for cleaving Aβ from its precursor APP (22). Another antiamyloid approach seeks to increase clearance of Aβ by active immunization with Aβ or passive immunization with anti-Aβ antibodies (23,24).
Given the increasing incidence of AD with age, coupled with the aging of the population, AD threatens to become one of the most significant public health concerns of the 21st century. The economic implications of increased AD prevalence have motivated a vigorous effort to identify potential new treatments for AD. Novel technologies capable of identifying subjects destined to develop AD prior to the onset of clinical symptoms will prove to be invaluable, as therapeutic interventions most likely will be optimally effective in the preclinical phase of the disease prior to irreversible neurological damage. Imaging technologies capable of noninvasively quantifying the hallmark characteristics of AD, amyloid plaques and NFTs in the brain, using positron emission tomography (PET) or single-photon emission computed tomography (SPECT) show promise as potential biomarkers for brain amyloidosis.
Motivation for Amyloid Imaging
A technique able to localize and quantify amyloid deposition in the living brain would represent a major advancement in the study of the pathophysiology and treatment of AD. A logical application for such a tool would be to critically assess the amyloid cascade hypothesis in longitudinal studies of at-risk, presymptomatic subjects (e.g., elderly controls and carriers of mutations causing eoFAD). Amyloid imaging might also provide key information that could help predict which MCI patients would and would not progress to AD. This information could provide a key piece of information that would allow clinicians to identify patients most likely to benefit from intervention with an antiamyloid therapeutic.
A third important application of such a tool would be to accelerate the development of antiamyloid therapies by providing quantitative observations of brain amyloid burden pre- and posttreatment. This would provide rapid feedback regarding the efficacy of candidate therapies, which could streamline efforts by focusing on the rapid development of the most promising therapies.
Although amyloid imaging agents are relative newcomers to the armamentarium of molecular imaging agents, they are beginning to provide some valuable (and sometimes unexpected) information that is helping to elucidate the mechanisms of AD and other dementias. This chapter summarizes what has been learned to date in this short but exciting journey.
Tracers for PET Amyloid Imaging Studies in Humans
Radiolabeled Monoclonal Antibody Fragments
The earliest documented attempts to image amyloid deposits in the brains of patients with AD using noninvasive imaging techniques employed a monoclonal antibody fragment, 10H3, which was targeted to the first 28 amino acids of the Aβ peptide. Labeled with technetium-99m (99mTc) for SPECT imaging, [99mTc]-10H3 exhibited some promising preclinical results, as it was demonstrated to bind significantly to neuritic plaques and cerebrovascular amyloid deposits on autoradiographic sections of human brain (25,26). However, systemic injection of [99mTc]-10H3 showed negligible brain entry that could not distinguish AD from control subjects (27). It is likely that the relatively large molecular weight of the agent contributed to low brain entry. This was not an altogether unexpected result, as poor blood–brain barrier penetration is a common feature of large molecular weight radiolabeled antibody fragments that limits their utility for molecular imaging of the brain (28). Although it may be possible to achieve detectible brain concentrations of radiolabeled antibody fragments after a protracted period of uptake of several days, the short biological half-life of 10H3 and the relatively short half-life of 99mTc (6 hours) rendered this approach impractical for visualizing brain amyloid deposits in vivo. Subsequent attempts to image amyloid in vivo focused on lipophilic small molecules capable of readily penetrating the blood–brain barrier.
Fluorine-18-FDDNP
A radiofluorinated derivative (termed FDDNP; Fig. 14.4.1) of the solvent- and viscosity-sensitive fluorophore 2-[1-[6-(dimethyl- amino)-2-naphthyl]ethylidene]malononitrile (DDNP) was the first PET agent to be investigated in humans for the purpose of visualizing amyloid in vivo. Agdeppa et al. (29,30) reported that FDDNP bound to both amyloid plaques and NFTs in vitro, and FDDNP was shown to bind with high affinity (Kd = 0.12 nM) to synthetic Aβ(1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40) fibrils using a fluorescence assay method (29). No binding assays using radioligand binding assay methods or cortical brain homogenates from pathologically confirmed AD subjects have been reported with FDDNP, nor have binding affinities for FDDNP to tau or NFTs been reported to date. Initial human studies of fluorine-18-labeled FDDNP ([18F] FDDNP) in AD patients (n = 9) and controls (n = 7) showed greater retention in frontal, parietal, temporal, and occipital cortices at steady state (60 to 120 minutes postinjection), although this increased cortical retention was shown to exceed the reference region (pons) by only 10% to 15% (31). The area of highest retention at equilibrium was the mesial temporal cortex (MTC), including hippocampus, amygdala, and
entorhinal cortex, and retention of [18F]-FDDNP in the MTC was shown to exceed retention in the pons by 30%. The mesial temporal cortex is a region known to be relatively free of neuritic plaques, but relatively enriched in NFTs compared to cortex (9,32).
entorhinal cortex, and retention of [18F]-FDDNP in the MTC was shown to exceed retention in the pons by 30%. The mesial temporal cortex is a region known to be relatively free of neuritic plaques, but relatively enriched in NFTs compared to cortex (9,32).
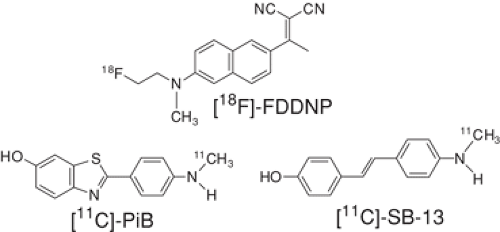 Figure 14.4.1. Structures of three PET amyloid imaging agents whose properties in human brain imaging studies have been reported. Radiofluorinated fluorophore 2-[1-[6-(dimethylamino)-2-naphthyl]ethylidene]malononitrile (FDDNP) is radiolabeled with fluorine-18 (110-minute half-life), while Pittsburgh compound-B (PiB) and stilbene derivative 4-N-methylamino-4′-hydroxystilbene (SB-13) are radiolabeled with carbon-11 (20-minute half-life). |
Initial analyses of [18F]-FDDNP PET data were performed using a novel kinetic analysis method, termed the relative residence time (RRT), which equates specific binding of radiotracer to the negative net difference between the reciprocal of the tissue clearance constants (k2) for the reference and amyloid-laden tissues. Although the authors demonstrated significantly longer RRT values for AD patients compared to controls, RRT values appeared to be sensitive to both peak and steady-state levels of [18F]-FDDNP in tissue. (31) This dependence on peak and steady-state radiotracer concentrations may result in an untoward influence of blood flow and transport phenomenon on the overall [18F]-FDDNP outcome measure. RRT estimates differed by a factor of 15 or more in brain regions with identical late-time levels of tracer, such as temporal and occipital cortex, indicating that it was the difference in peak entry levels into these brain areas that was driving the difference in RRT.
The applicability of conventional quantitative analysis methodologies, such as compartmental modeling and graphical analyses, to dynamic [18F]FDDNP PET data remains to be demonstrated, although a recent report employed the noninvasive Logan graphical analysis (33) without demonstrating attainment of steady-state conditions (34). These studies are necessary to fully understand the nature of [18F]-FDDNP retention and to assess the overall sensitivity and specificity of the compound for the detection of AD pathology in vivo.
Carbon-11-Pittsburgh Compound-B
A carbon-11-labeled benzothiazole derivative of the amyloid dye thioflavin-T, termed Pittsburgh compound-B (PiB; Fig. 14.4.1), was the second PET agent to be employed in human in vivo amyloid imaging studies (35,36). Tritiated-PiB was shown to bind specifically and with high affinity (Kd = 1.4 nM) to frontal cortex brain homogenates from pathologically confirmed AD subjects, while cerebellar and white matter tissue did not bind [3H]-PiB specifically (37). In addition, [3H]-PiB bound to synthetic Aβ(1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40) fibrils with a similar high affinity (Kd = 4.7 nM). The initial human study of [11C]-PiB by Klunk et al. included 16 patients with mild AD (minimental state examination [MMSE] scores of 18 to 28) and nine healthy control subjects. Compared to controls, AD patients showed greater than twofold higher retention of [11C]-PiB in areas of brain association cortex known to contain large amounts of amyloid deposits in AD (P <.002), but the levels of retention in areas known to be relatively unaffected by amyloid pathology (e.g., subcortical white matter, pons and cerebellum; P >.2) were equivalent.
Later investigations built on this proof-of-concept study and aimed to extend these observations to include longer periods of data acquisition (90 minutes), coregistered magnetic resonance images for precise region-of-interest definition, and arterial input function determination. These refinements sought to identify a valid and robust compartmental model that described the in vivo kinetics of [11C]-PiB across the spectrum of control and AD subjects. These fully quantitative analyses concluded that the [11C]-PiB PET data were well described by a two-tissue four-parameter (2T-4k) compartmental model that assumed reversible in vivo kinetics and negligible occupancy of the binding sites by the radiotracer or carrier. These studies also identified the distribution volume ratio (DVR) as the preferred outcome measure, as it provided a reliable nonnegative index of amyloid deposition in both AD and control subjects (38).
Although the 2T-4k model was found to best describe [11C]-PiB kinetics in vivo, difficulties in estimating small parameter values in regions with little or low signal (i.e., control brain or cerebellum) resulted in spuriously overestimated DVR values and hence higher test-retest variability. For this reason, the Logan graphical analysis (39) using 90 minutes of data (ART90) was investigated as a potential alternative to the 2T-4k analysis. ART90 methods showed significantly lower intersubject variability than the 2T-4k analysis (generally 10% to 20%), although DVR measures obtained using the two methods were strongly correlated (r ∼0.9) across all regions of interest.
ART90 DVR measures were found to be approximately twofold higher in cortical regions of AD subjects, relative to controls. These findings, coupled with an observed test-retest variability of approximately 7% across regions, identified ART90 as the optimum arterial-input–based method for human [11C]-PiB studies and the method against which later simplified methods of analysis would be compared.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


