Pulmonary Arterial Hypertension
In 2003 in Venice, the World Health Organization adopted a revised classification for pulmonary arterial hypertension (Table 12.1).
The majority of the causes of pulmonary arterial hypertension included in the World Health Organization classification have been covered in previous chapters. In this chapter, idiopathic or primary pulmonary hypertension is illustrated as a chronic cause of pulmonary arterial hypertension. In addition, some of the acute causes of pulmonary arterial hypertension and their consequences are illustrated with exercise-induced pulmonary hemorrhage and high-altitude pulmonary edema.
 Primary Pulmonary Hypertension
Primary Pulmonary Hypertension
 By definition, primary pulmonary hypertension is pulmonary arterial hypertension without a known cause.
By definition, primary pulmonary hypertension is pulmonary arterial hypertension without a known cause.
 Pulmonary arterial hypertension is defined as a mean pulmonary artery pressure exceeding 25 mm Hg (3300 pascals [Pa]) at rest or 30 mm Hg (4000 Pa) with exercise.
Pulmonary arterial hypertension is defined as a mean pulmonary artery pressure exceeding 25 mm Hg (3300 pascals [Pa]) at rest or 30 mm Hg (4000 Pa) with exercise.
 The pathologic changes are largely confined to the muscular pulmonary arteries that measure less than 1 mm in diameter. The changes seen in large pulmonary arteries are a response to the pulmonary hypertension.
The pathologic changes are largely confined to the muscular pulmonary arteries that measure less than 1 mm in diameter. The changes seen in large pulmonary arteries are a response to the pulmonary hypertension.
 The histologic features range from minor changes of hypertrophy of the muscular media of the small pulmonary arteries to subendothelial fibrous proliferation and “plexiform lesions.” These pathologic changes are seen in both primary pulmonary hypertension and pulmonary hypertension with known causes.
The histologic features range from minor changes of hypertrophy of the muscular media of the small pulmonary arteries to subendothelial fibrous proliferation and “plexiform lesions.” These pathologic changes are seen in both primary pulmonary hypertension and pulmonary hypertension with known causes.
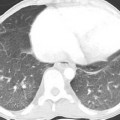
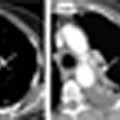
 Radiologic abnormalities include dilatation of the pulmonary arteries (Fig. 12.1).
Radiologic abnormalities include dilatation of the pulmonary arteries (Fig. 12.1).
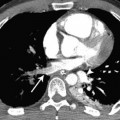
 As a result of chronically raised pulmonary artery pressure, atheromatous changes may develop in the pulmonary arteries, which can calcify (Fig. 12.2).
As a result of chronically raised pulmonary artery pressure, atheromatous changes may develop in the pulmonary arteries, which can calcify (Fig. 12.2).
 On high-resolution computed tomography (CT), subtle centrilobular ground-glass opacities are often noted (Fig. 12.1), which correspond to cholesterol granulomas. These possibly form as a consequence of macrophage ingestion of red blood cells after repeated pulmonary hemorrhage.
On high-resolution computed tomography (CT), subtle centrilobular ground-glass opacities are often noted (Fig. 12.1), which correspond to cholesterol granulomas. These possibly form as a consequence of macrophage ingestion of red blood cells after repeated pulmonary hemorrhage.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


