Arthritis is the most commonly recognized manifestation of rheumatic disease, but pulmonary manifestations can occur in nearly all disorders and are found in various age groups in children. Symptoms of pulmonary disease in children may be subtle or absent, and children at elevated risk should be screened for pulmonary involvement.
The combination of well-developed vascularity, exposure to external stimuli and toxins in ambient air, and a covering (the pleura) that shares structures and functions analogous to joint synovia contributes to the susceptibility of the lungs to rheumatic and vasculitic inflammatory expression. Although the mechanisms of the pulmonary inflammation of rheumatologic disorders differ by etiology, common patterns of sequence and expression are seen. The initial inflammatory lesions usually lack an identifiable etiology and behave as though the child’s pulmonary tissue is foreign (hence the term “autoimmunity”). Genetic susceptibility or genetic modifiers are now well established for many disorders. Although acute expression with resolution sometimes occurs, chronic and relapsing scenarios are more common. The general sequence is as follows:

In some disorders, such as systemic lupus erythematosus (SLE), bleeding and infection are integral participants in the expression of the chronic inflammation. The cycle of immune-mediated inflammation, vascular leakage, and attempts at repair recurs with progressive organ damage, leading to loss of organ function. Available pharmacologic therapies directed at alleviating the inflammation in these disorders inhibit, but do not yet arrest the inflammatory process enough to prevent further organ damage.
The list of childhood rheumatologic disorders is long. This chapter reviews the most common conditions and their associated pulmonary manifestations.
Systemic Lupus Erythematosus
SLE is an autoimmune disease characterized by multisystem inflammation and tissue damage. It commonly develops in young women, but patients of either gender or any age may be affected. In pediatric populations, SLE prevalence is similar in prepubescent boys and girls, but female predominance increases steadily after puberty. The prevalence of SLE increases with age, and SLE can develop in individuals of any race or ethnicity. Neonatal lupus occurs rarely and is caused by the passage of autoantibodies (anti-Ro, SSA and anti-La, SSB) across the placenta that cause congenital heart block or a self-limited lupus rash that manifests shortly after delivery and persists until maternal antibodies disappear, usually in the first year of life.
Pulmonary manifestations are common. Excluding common pulmonary infections, the pleura is the most frequently involved tissue of the pulmonary system. Other parenchymal complications—acute lupus pneumonitis (ALP), alveolar hemorrhage (AH), pulmonary embolism, and pulmonary arterial hypertension (PAH)—occur less frequently, but are potentially lethal. Although the clinical literature of SLE is focused on adult studies, the reported pediatric literature suggests similar arrays of pulmonary manifestations and clinical syndrome courses. The prevalence of pulmonary involvement is difficult to estimate because of selection bias related to methodologies that are retrospective or case-series in nature. Pediatric data are insufficient to draw inferences regarding the variation in disease expression based on age or physical development. A large histopathologic study of lung involvement with SLE in children suggests that these lesions do not differ from the lesions in adults. These and other studies confirm that the more uncommon cases of chronic lung disease develop in children as well, including symptomatic chronic interstitial lung disease (ILD) and shrinking lung syndrome. Drug-induced lupus (DIL) is often characterized by lung involvement. Infection remains a leading cause of death in patients with SLE, however. In view of the risk of pulmonary infection associated with immunosuppressive treatments for SLE, the clinical approach to patients with pulmonary complaints should always include a thorough search for infection. Initial treatment regimens for acutely ill patients should incorporate empiric antibiotic therapy until the precise cause of the pulmonary disease is ascertained.
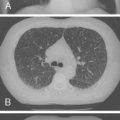
Noninfectious pulmonary complications of SLE are summarized in Table 10-1 . Pleuritis is the most common pulmonary manifestation of SLE. Symptoms include pleuritic chest pain and dyspnea in association with other features of lupus disease activity, such as fever, fatigue, rash, and arthritis. Chest radiographs may show small to moderate pleural effusions. Large effusions are unusual ( Figs. 10-1 and 10-2 ). When pleural effusion is noted in SLE, diagnostic thoracentesis for pleural fluid aspiration and analysis is indicated. These effusions are typically serous or serosanguineous exudates. Hemorrhagic effusions associated with hypoxemia can occur with pulmonary infarcts and infection. These entities should always be considered in the differential diagnosis of pleuritis, particularly in the absence of coexisting features of systemic lupus activity. Pleural pathology in SLE consists of local inflammation with immunoglobulin and complement deposition. In some cases, pleural fibrosis is observed, although the fibrosis is infrequently considered clinically important.
| PULMONARY MANIFESTATION | ESTIMATED PREVALENCE | ONSET | PRESENTING SIGNS AND SYMPTOMS | RADIOGRAPHIC CHANGES | PROGNOSIS | TREATMENT | DIFFERENTIAL DIAGNOSIS |
|---|---|---|---|---|---|---|---|
| Pleuritis | 50–80% | Acute | Pleurisy, dyspnea, orthopnea, pleural rub | Pleural effusion | Good | NSAIDs, corticosteroids | Infection, PE, ALP, AH, PTX, drug toxicity |
| ALP | ≤10% | Acute | Dyspnea, cough, fever, chest pain, pleurisy, hemoptysis; often preceded by or associated with infection, hypoxemia | Patchy acinar infiltrates with bibasilar predominance | Poor; mortality 50% | Corticosteroids, plasmapheresis, CYC, AZA | Infection, PE, AH, pleuritis |
| AH | ≤2% | Acute | Dyspnea, cough, chest pain, decrease in hemoglobin, pleurisy, hypoxemia | Patchy acinar infiltrates with bibasilar predominance | Mortality 50% | Corticosteroids, plasmapheresis, CYC, AZA | Infection, ALP, PE, vasculitis, Goodpasture syndrome, IPH |
| Acute reversible hypoxemia | <1% | Acute/chronic | Dyspnea, low FVC, low DLCO | Normal | Good | Corticosteroids | Infection, PE, early ALP or AH, shrinking lung syndrome |
| Chronic interstitial pneumonitis | 3% | Chronic, may be long-term complication of ALP | Dyspnea, cough, low FVC, low DLCO, fibrosis | CXR—reticular interstitial infiltrates: ground glass, honeycombing | Poor to good | Corticosteroids, CYC, AZA | Infection, LIP, drug toxicity, chronic aspiration |
| Shrinking lung disease | <1% | Chronic | Dyspnea, orthopnea, low FVC, low DLCO | Normal or basilar atelectasis, elevated diaphragm | Good | Corticosteroids | Respiratory muscle weakness from myopathies |
| Thromboembolic disease | — | Acute | Dyspnea, pleurisy, hemoptysis, fever, pleural rub | CXR—normal or effusion | Variable | Anticoagulation | Infection, pleurisy, PTX, AH or ALP |
| Pulmonary hypertension | 5–14% | Chronic | Dyspnea, chest discomfort, right heart failure, pericardial effusion, elevated BNP, low DLCO with stable FVC | CXR—normal or perfusion | Variable | Pulmonary vasodilators (ETRA, prostanoids, phosphodiesterase type 5 inhibitors, anticoagulants; CYC and corticosteroids in select cases | Infection, interstitial lung disease |




Mild pleuritis is usually treated effectively with nonsteroidal anti-inflammatory drugs (NSAIDs). In more severe cases, such as large symptomatic effusions, corticosteroids are indicated. If the pleural effusions are not associated with significant multisystem activity, efforts should be made to limit corticosteroid exposure, especially in children, by using short pulses of therapy instead of prolonged tapers. Recurrent episodes of pleurisy may respond to treatment with hydroxychloroquine or methotrexate. More aggressive immunosuppressants are needed infrequently.
ALP is a rare complication of SLE, commonly manifesting with an acute, profound loss of respiratory function. ALP occurs in 1% to 4% of patients and accounts for nearly 4% of SLE hospital admissions. Although ALP may be a forme fruste of SLE, it most frequently occurs in patients with established disease. The clinical presentation includes acute onset of dyspnea, cough, fever, and hypoxemia. Coexisting infection is a common observation, suggesting that bacterial or viral infections may trigger ALP. Many patients report pleuritic chest pain. Hemoptysis occurs occasionally, but less frequently than the AH syndrome of pulmonary lupus. Chest radiographs show a diffuse or patchy acinar infiltrate with lower lobe predominance. With a rapidly progressive respiratory decline, many patients require mechanical ventilation. Bronchoscopy is suggested to exclude underlying infection, but because of the frequent coexistence of pulmonary infection with ALP, the discovery of a pathogen does not obviate the need for aggressive immunosuppressive therapy. Culture and antimicrobial sensitivity testing of bronchoscopic or open lung biopsy specimens is helpful in directing antibiotic therapy.
The histopathologic features of ALP appear similar to diffuse alveolar damage. Inflammatory cellular infiltrates involve the interstitium and alveolar wall. Nonspecific features, including edema, hemorrhage, and hyaline membranes, are commonly observed. Complement and immunoglobulin deposition may be shown by direct immunofluorescent staining, but vasculitic lesions are uncommon.
Treatment of ALP is based primarily on retrospective analyses of clinical cohorts, case series, and anecdotal reports. Typical treatment incorporates high dose, intravenous corticosteroids (1 to 2 mg/kg/day of prednisone or equivalent, usually to a maximum of 60 mg/day) and supportive respiratory care. Because mortality rates approach 50%, rapidly progressive respiratory decline indicates a need for more aggressive interventions, such as pulse intravenous methylprednisolone (30 mg/kg/day up to 1000 mg/day for 3 days) and plasmapheresis. Additional immunosuppressive agents, including azathioprine and cyclophosphamide, should be considered in patients who have persistent or recurrent disease. Some centers advocate early initiation of azathioprine or cyclophosphamide in patients who are seriously ill.
ALP is usually expressed as a severe, but self-limited, monophasic inflammation. Chronic interstitial disease may rarely develop in survivors of the acute illness, however. The incidence of chronic progression of ALP seems to be low enough that prolonged corticosteroid or immunosuppressive use is not usually necessary in patients who recover quickly from the initial onset of ALP. These patients should be monitored closely for pulmonary decline by determining serial pulmonary function every 3 to 6 months.
AH in SLE occurs rarely, but it may account for 3.7% of all SLE-related hospitalizations. Although AH can be the initial manifestation of SLE, most patients have an established diagnosis. AH most often occurs in patients who have high anti-dsDNA titers. Coexisting renal disease is seen in 60% to 90% of patients with AH. Most patients are young women who present with dyspnea, sometimes fever and pleurisy, and acute hypoxemia. Frank hemoptysis occurs in more than 50% of patients. Most patients with AH exhibit hemoptysis at some point during their illness, helping to differentiate AH from ALP. Pulmonary bleeding is usually substantial with an average hemoglobin decrease of approximately 7%. Chest radiographs show diffuse, bilateral acinar infiltrates that may progress rapidly over hours and in some cases just as rapidly resolve. The respiratory compromise in more than half of patients is rapid and profound, resulting in a need for supplemental oxygen and assisted ventilation. With assisted ventilation, the addition of positive end-expiratory pressure should theoretically be beneficial, impeding alveolar bleeding and maintaining alveolar expansion. Occasional cases of mild AH may be managed more conservatively. Because of the propensity for acute decompensation, all suspected cases of AH should be managed with caution, preferably with hospitalization. Mortality rates with AH are 40% to 90% in published series, with many fatalities occurring early.
Pulmonary infection may coexist in some cases. The relevance of infection as a causal agent is uncertain. The frequency of coexisting infection makes bronchoscopy a useful approach to the evaluation of these patients, however. As in ALP, the presence of a pulmonary pathogen in bronchoalveolar lavage (BAL) or biopsy specimens does not obviate the need for immunosuppression, but it helps guide the choice of antibiotics. Most patients have evidence of frank hemorrhage on BAL. In patients without bloody lavage fluid, the presence of hemosiderin-laden macrophages is evidence of recent hemorrhage. Bronchoscopic and open lung biopsy specimens show nonspecific interstitial and alveolar wall infiltrates of lymphocytes and neutrophils, edema, and hyaline membranes. Hemosiderin-laden macrophages or inflammation of small pulmonary arterioles or capillaries (known as capillaritis) helps distinguish AH from ALP. Direct immunofluorescent staining may show granular interstitial or endothelial staining for complement and immunoglobulins. This differentiates AH from Goodpasture syndrome, in which the histopathology is characterized by a pattern of linear immunofluorescence.
The prognosis of AH is similar to ALP and relates proportionately to the severity at presentation. Treatment recommendations are based on case series and anecdotal reports, although the early use of plasmapheresis in severe cases is based on the success of this therapy in Goodpasture syndrome, which is similarly characterized by recurrent AH. The core initial therapy is aggressive intravenous corticosteroid dosing (1 to 2 mg/kg/day of prednisone or equivalent) or may begin with intravenous pulse methylprednisolone (1000 mg/day × 3 days). Early initiation of cyclophosphamide is advocated by some centers. Even the addition of this potent immunosuppressant is not uniformly effective at reversing respiratory failure, however. Some patients hemorrhage recurrently at variable intervals. Plasmapheresis may be indicated in patients who have recurrent hemorrhages even after starting corticosteroids because this may provide a more rapid onset of immunosuppression than cyclophosphamide. Nonetheless, efficacy data for plasmapheresis therapy are lacking. Even the published data for cyclophosphamide are conflicting. Some analyses have suggested that cyclophosphamide use is a risk factor for AH-associated mortality; however, these results probably reflect referral bias because cyclophosphamide is more likely to be used in patients who are more seriously ill.
The frequency and range of expression of chronic ILD in patients with SLE is not well documented. Chronic ILD probably affects less than 2% of lupus patients, and many of these patients probably have clinically mild illness. Some authors have reported chronic ILD developing as a consequence of ALP. In some cases, interstitial scarring and restrictive changes on pulmonary function tests (PFTs) probably reflect injury from the acute pulmonary inflammation. Progressive dyspnea and pulmonary function decline in a subset of patients, however, probably reflecting more classically chronic progressive ILD. Reports from other authors have described a few patients with chronic ILD presenting typically with cough, dyspnea, radiographic interstitial infiltrates or honeycomb changes, and restrictive lung disease pattern on PFTs. Histologic data are sparse, but include nonspecific interstitial inflammation with changes of fibrosis. Some patients with chronic ILD apparently respond to corticosteroids, rarely with marked improvement.
In our center, we have observed patients with two different histologic patterns of ILD. A young African-American woman with a history of antiphospholipid antibody syndrome presented with persistent dyspnea and cough over 12 months. Repeated high-resolution computed tomography (CT) scans performed during this period showed transient patchy interstitial infiltrates. Initially, the concern was for recurrent pulmonary emboli, even though she was being aggressively anticoagulated. Lung tissue obtained by video-assisted thoracoscopy open lung biopsy showed changes consistent with nonspecific interstitial pneumonitis, however, without any evidence of thrombosis or of fibroblastic foci or honeycomb changes. This patient was treated with oral corticosteroids for several months. Her respiratory symptoms and scan findings completely resolved.
In a second case, an older white woman with inflammatory arthritis and subacute cutaneous lupus presented with progressive dyspnea, chronic cough, and restrictive lung disease pattern on PFTs. High-resolution CT scan revealed patchy ground-glass findings and honeycomb changes with a peripheral and basilar predominance. A lung biopsy performed by video-assisted thoracoscopy revealed changes consistent with nonspecific interstitial pneumonia with fibroblastic foci. In addition, areas of heterogeneous honeycomb scar were noted on lower lobe specimens. The patient was treated with corticosteroids and 12 months of intravenous pulse cyclophosphamide because the lung pathology was most characteristic of ILD seen in scleroderma (systemic sclerosis). Substantial improvements in respiratory function (including elimination of her initial dependence on supplemental oxygen) in addition to reductions in ground-glass opacities on scanning were observed. After a course of cyclophosphamide, the patient was transitioned to oral azathioprine for maintenance therapy and has not experienced relapse of ILD after 1 year.
Although chronic forms of ILD are rare in SLE, they occur occasionally and may be misdiagnosed as manifestations of SLE. Careful evaluation, including serial PFTs and using prone imaging, can increase diagnostic sensitivity. Most patients should undergo open lung biopsy to confirm the diagnosis; determine the degree of fibrosing interstitial disease; and exclude other pathology, such as thrombosis, malignancy, and chronic infection. Treatment may be limited to a course of corticosteroids in patients with pathology limited to nonspecific interstitial pneumonitis, but prolonged courses of cyclophosphamide, methotrexate, or azathioprine should be considered in patients with changes of fibrosis on biopsy.
Patients with SLE are at risk for the development of venous thrombosis and thromboembolism. Approximately 30% to 40% of patients with SLE have circulating antiphospholipid antibodies. These antibodies are associated with an increased risk for venous and arterial thrombosis and pregnancy wastage, particularly late first-trimester and second-trimester pregnancy loss. In patients with antiphospholipid syndrome (defined as the presence of anticardiolipin antibodies or lupus anticoagulant and thrombosis or defined pregnancy loss), recurrent thrombosis is common. Treatment following a thrombosis requires lifelong anticoagulation, and subsequent pregnancies after a loss should be managed with heparin through 3 to 6 months postpartum.
Venous thrombosis and thromboembolism are the most common thrombotic complications of antiphospholipid antibody syndrome. Lupus patients presenting with acute dyspnea, with or without pleurisy or hemoptysis, should undergo immediate evaluation for pulmonary embolus, including ventilation/perfusion or spiral CT and lower extremity Doppler ultrasound to detect deep vein thrombosis. The absolute risk for clot in patients with antiphospholipid antibodies and no prior history of thrombosis is unknown. Procoagulant risk factors, including cigarette smoking, estrogen-based contraceptive use, and nephrotic-range proteinuria from renal disease, likely increase the risk. In rare cases, chronic thromboembolism can lead to the development of PAH. In patients presenting with PAH complicating SLE, careful ventilation/perfusion scanning is necessary to exclude chronic pulmonary thromboembolic disease.
PAH is not as rare in SLE as previously thought, and is characterized by progressive dyspnea and hypoxemia that progresses rapidly to right heart failure. PAH may be an idiopathic condition typically occurring in healthy young women or may develop secondary to other conditions, including autoimmune disease (most commonly seen in scleroderma), chronic venous thromboembolism, congenital heart defect resulting in arterial-to-venous shunts, human immunodeficiency virus (HIV), sickle cell disease, prior history of anorexigenic weight loss drugs, and portopulmonary disease of liver failure. Originally considered a rare SLE complication, PAH is now recognized to develop in 1% to 14% of patients. As with scleroderma patients, lupus-associated PAH has a poor prognosis, with mortality rates of 50% within 2 years of diagnosis.
PAH often is unrecognized in early stages. Patients typically present with dyspnea on exertion. By the time more alarming signs and symptoms develop, most patients have progressed to substantial, and in some cases irreversible, right heart failure. Risk factors for the development of PAH in SLE patients are not well defined, although a greater percentage of these patients with PAH report Raynaud phenomenon and have measurable antiphospholipid antibodies. Significant right heart failure is suggested by a constellation of any of the following signs: syncope, ascites, lower extremity edema, right ventricular heave, or a right-sided S 3 auscultatory sound. Careful evaluation of diagnostic testing increases the sensitivity for detecting early PAH. Echocardiogram is the primary screening tool and should be performed in all SLE patients presenting with dyspnea. Estimated peak right ventricular pressures are often elevated. Signs of right heart strain, such as right atrial or ventricular enlargement and dyskinetic septal motion, strongly suggest PAH, but are more common in advanced disease. PFTs may show a disproportionate decrease in diffusing capacity for carbon monoxide (DLCO) compared with decreases in lung volumes. In scleroderma patients, serum brain natriuretic peptide seems to be a sensitive and specific marker of PAH in patients with normal left ventricular function. Histopathologic remodeling in PAH in SLE patients is nonspecific. Vascular lesions include intimal thickening, medial smooth muscle hypertrophy, and plexiform pathology, lesions also seen in scleroderma and idiopathic PAH.
The diagnosis of PAH is suggested by dyspnea (in the absence of an alternative cardiopulmonary explanation) in conjunction with an elevated peak right ventricular systolic pressure on echocardiogram, accompanied by possible elevations of the forced vital capacity (FVC)-DLCO ratio and serum brain natriuretic peptide levels. The confirmatory diagnostic test that is considered a gold standard is a right heart catheterization. The catheterization findings outlined in Table 10-2 confirm the diagnosis of PAH. Because of the co-occurrence of shunts resulting from congenital heart disease in children, an experienced pediatric cardiologist or critical care specialist should perform the right heart catheterization. Echocardiograms are insensitive instruments to detect shunts; a careful right heart catheterization is crucial in all patients with suspected PAH.
| RIGHT HEART CATHETERIZATION MEASUREMENT | RESULT CONFIRMING PULMONARY ARTERY HYPERTENSION * |
|---|---|
| Elevated mean pulmonary arterial pressure | >25 mm Hg at rest (>30 mm Hg with exercise) |
| Normal pulmonary capillary wedge pressure | <15 mm Hg |
| Elevated pulmonary vascular resistance | >3 Wood units |
* All features are necessary to confirm diagnosis of pulmonary artery hypertension.
When SLE patients are diagnosed with PAH, chronic venous thromboembolic disease should be carefully considered. Standard treatment of all SLE patients with PAH includes anticoagulation, but the level of anticoagulation may need to be increased for patients with antiphospholipid antibodies who have developed clots. Treatment for SLE-associated PAH differs from treatment of PAH in patients with scleroderma. In the scleroderma group, no benefit has been observed from the use of immunosuppressive therapy. Evidence from case reports and retrospective studies in SLE suggest, however, that a subset of patients improve or experience reversal of PAH with aggressive immunosuppressive therapy incorporating corticosteroids and cyclophosphamide.
In our adult rheumatology clinic, all patients with SLE who develop PAH, excluding PAH resulting from congenital heart disease or chronic venous thromboembolic disease, are treated with six monthly doses of intravenous pulse cyclophosphamide (1 g/m 2 /dose) and tapering doses of corticosteroids. Concomitant treatment with standard PAH drugs is initiated in all patients with significant clinical symptoms (World Health Organization class III and IV) or with 6-minute walk distance test less than 320 meters. In patients with mild disease (World Health Organization class I and II) who have good 6-minute walk distance test, PAH drugs may be held while the patient is treated with immunosuppression, provided that the patient is followed carefully with monthly visit, quarterly echocardiograms, and 6-minute walk tests.
Shrinking lung syndrome is a rare complication of SLE, although it is possible that mild cases may go undiagnosed. Patients characteristically present with progressive dyspnea and a restrictive lung pattern on PFTs. Orthopnea is a unique clinical feature of shrinking lung syndrome. DLCO may or may not be decreased. Chest radiographs reveal elevated diaphragms and bibasilar atelectasis. This finding may be present, however, even when there is no evidence of parenchymal or pleural disease. The differential diagnosis includes ILD; this does not show inflammatory or fibrotic changes, however. ILD is not a feature of shrinking lung syndrome. The cause of shrinking lung syndrome is likely to be multifactorial. In a few cases, progressive pleural fibrosis results in restrictive lung disease. More recent studies suggest that at least some patients have reduced lung volumes secondary to diaphragmatic and intercostal muscle weakness.
Although this condition was previously considered relentlessly progressive, most cases of shrinking lung syndrome seem to have a good prognosis, stabilizing or sometimes spontaneously improving over time. For symptomatic patients, a trial of corticosteroids is indicated, although the response rates from limited published data are uncertain. Occasionally, progressive dyspnea and loss of lung volumes requires cytotoxic treatments, such as azathioprine or cyclophosphamide. Little is known about shrinking lung syndrome in pediatric SLE, with only a few cases reported. In our clinic, we care for a boy with shrinking lung syndrome and concomitant valvulitis requiring a valvular replacement.
The airways are infrequently affected in SLE ( Table 10-3 ). Rare cases of bronchiolitis obliterans with organizing pneumonia (BOOP) have been reported. BOOP lesions are characterized histologically by inflammatory changes within the distal airways and alveoli associated with nonspecific bronchiolar inflammation. Patients present with dyspnea caused by a restrictive ventilatory deficit. Fever, interstitial infiltrates, and cough may be part of the initial presentation. In most cases, open lung biopsy is needed to confirm the diagnosis. Treatment typically is initiated with corticosteroids, which are usually effective. The etiology of BOOP in SLE is uncertain, but may be induced by infection.
| PATHOLOGY | ESTIMATED PREVALENCE | |
|---|---|---|
| Upper airway | Laryngeal inflammation | Rare |
| Epiglottitis | Rare | |
| Subglottic stenosis | Rare, may be risk in postextubation period | |
| Lower airway | Reactive airway disease | Rare, may be associated with reflux and aspiration in patients with esophageal dysmotility |
| Bronchiolits obliterans with organizing pneumonia | Probably rare |
Primary upper airway disease in SLE is extremely rare. A few authors have suggested an increased risk for postintubation subglottic stenosis. Extra care may be required for SLE patients after extubation. Obstructive lung disease is uncommon in SLE. Some patients develop esophageal dysmotility, however, similar to that seen in scleroderma. As a result, the lower esophagus can become patulous, resulting in an increased risk for aspiration, particularly at night when the patient sleeps prone. Common symptoms of aspiration include nocturnal cough and wheezing. PFTs may show a decreased forced expiratory volume in 1 second (FEV 1 )/FVC ratio. A decreased DLCO in the setting of preserved lung volumes also may suggest chronic aspiration. Esophageal dysmotility is often evident on barium swallow, although incidental detection of a patulous esophagus may occur with chest CT scans ordered to exclude ILD. High-resolution CT may show patchy ground-glass opacities in the right lower lobe. Treatment includes aggressive acid-reducing therapy, preferably with proton-pump inhibitors. In addition, antiaspiration interventions ( Table 10-4 ) and prebedtime doses of metoclopramide to enhance lower esophageal and gastric emptying may benefit some patients.
| PATHOLOGY | INTERVENTION |
|---|---|
| Acid reflux and aspiration | Proton-pump inhibitor; may require dose titration to eradicate symptoms of heartburn |
| Aspiration | Raise the head of the bed 2–4 inches |
| Avoid bed wedges, multiple pillows | |
| Nothing to eat or drink within 3–4 hr of going to bed | |
| Avoid caffeinated beverages after dinner | |
| Esophageal dysmotility | Prokinetic therapy, preferably metoclopramide, taken before bedtime; additional dose of metoclopramide before dinner may be necessary |
Patients with SLE have immunocompromise, placing them at risk for respiratory tract infection, often with opportunistic organisms. Compromised immune responsiveness in SLE is a result of abnormalities of complement or cell-mediated immunity, reduced pulmonary macrophage and phagocytic function, treatment with medications (corticosteroids and immunosuppressive agents), and compromised airway clearance resulting from airway inflammation and weak cough (respiratory muscle weakness). The most common organisms responsible for respiratory tract infection in SLE patients include Klebsiella aerobacter, Escherichia coli , and β-hemolytic streptococcus. Candida albicans, Aspergillus species, and Pneumocystis jiroveci (formerly Pneumocystis carinii ) also have been cultured from the lungs of these patients. Patients with immune dysfunction and patients receiving immunosuppressive therapy should receive routine trimethoprim-sulfamethoxazole prophylaxis.
Drug-Induced Lupus
The development of DIL syndromes has been most strongly associated with procainamide, hydralazine, and phenytoin. Through various mechanisms, these agents induce autoreactive immune responses in some patients, resulting in clinical lupus-like disease. Other agents have been associated with lupus-like conditions ( Table 10-5 ) that are more heterogeneous than classic DIL. Pleuropulmonary manifestations, particularly pleurisy and pleural effusions, are the most common clinical features of DIL; other features include arthralgia, fever, and, less commonly, rash. Most affected patients also develop autoantibodies, including antinuclear antibodies (ANAs) and antihistone antibodies. After discontinuation of the offending drug, the clinical manifestations are self-limited in most cases. Treatment is typically conservative, using NSAIDs and occasionally corticosteroids to manage symptoms.
| STRENGTH OF ASSOCIATION WITH DRUG-INDUCED LUPUS | AGENT | PLEUROPULMONARY MANIFESTATIONS (%) |
|---|---|---|
| Strong | Procainamide | 75 |
| Hydralazine | 25 | |
| Phenytoin | Probably uncommon | |
| Moderate | Isoniazid | Uncommon |
| Methyldopa | Uncommon | |
| Penicillamine | Uncommon | |
| Quinidine | Uncommon | |
| Minocycline | Uncommon | |
| Tumor necrosis factor-α inhibitors | Uncommon | |
| Weak | Statins | Uncommon |
| Interferons | Uncommon | |
| Terbinafine | Uncommon | |
| Zafirlukast | Uncommon |
Chronic SLE and internal organ involvement, particularly central nervous system or renal disease, are very unusual complications of DIL. Antihistone antibody titers often decrease as DIL wanes, but ANA titers may remain elevated indefinitely and are not worthy of serial assessment. In rare cases, usually with procainamide, pulmonary parenchymal disease may develop and even be associated with pulmonary fibrosis. Aggressive immunomodulatory therapy is not typically indicated, however, even in these settings. Because DIL may take weeks to months (rarely up to a year) to resolve, patients with pulmonary manifestations, particularly dyspnea or abnormal PFTs, should be monitored on a regular basis. Pulmonary manifestations with drugs less traditionally associated with DIL, such as minocycline, are less common and should be viewed as potential manifestations of another pulmonary process or evidence of chronic SLE that has been unmasked by the drug.
Mixed Connective Tissue Disease
Mixed connective tissue disease (MCTD) is categorized as an overlap syndrome because patients manifest features that are characteristic of multiple defined autoimmune disorders ( Table 10-6 ). They often develop a mixture of features of SLE, rheumatoid arthritis, and dermatomyositis. The defining serologic feature of MCTD is the presence of antiribonucleoprotein autoantibodies. Table 10-7 lists the most common clinical findings. In MCTD, serious complications of lupus, such as nephritis or central nervous system pathology, are unusual. Consequently, MCTD is generally regarded as having a better prognosis, provided that inflammatory muscle disease and deforming synovitis can be controlled with immunomodulatory therapy. Prospective cohort studies now suggest, however, that the insidious development of ILD or PAH results in a substantial increase in morbidity and mortality for a subset (approximately 20% to 30%) of patients with MCTD.
|
* Must meet all three criteria to be diagnosed with mixed connective tissue disease.
| CLINICAL FEATURE | FREQUENCY IN ADULTS (%) | FREQUENCY IN CHILDREN (%) |
|---|---|---|
| Inflammatory arthritis | 95 | 82–93 |
| Myositis | 63 | 47–61 |
| Pulmonary disease | 20–80 | 35–60 |
| Raynaud phenomenon | 85 | 85–94 |
| Skin rash | 38 | 33–38 |
| Esophageal dysmotility | 67 | 21–41 |
| Hepatosplenomegaly | 15–19 | 29 |
| Serositis | 43 | 23–28 |
| Antiribonucleoprotein antibody | 100 | 100 |
| Rheumatoid factor | 70 | 57–68 |
There are many reported pleuropulmonary manifestations of MCTD. ILD, pleuritis, and PAH are the most common and clinically important forms ( Table 10-8 ). Pleuritis occurs occasionally, a distinct difference from SLE, but rarely MCTD can be associated with large, symptomatic pleural effusions. Patients typically are managed with NSAIDs for mild to moderate pleuritis or corticosteroids for large symptomatic effusions. Rarely, venous thromboembolic disease associated with antiphospholipid antibodies may occur. Progressive esophageal dysmotility is far more common in MCTD than in SLE, and increases the risk for aspiration pneumonia or chronic aspiration–associated bronchoconstriction or mild pulmonary fibrosis. The management of antiphospholipid antibody syndrome and esophageal dysmotility in MCTD is identical to that in SLE patients.
| PLEUROPULMONARY DISEASE | FREQUENCY (%) | CLINICAL FEATURES | DIAGNOSTIC TESTING | DIFFERENTIAL DIAGNOSIS | TREATMENT |
|---|---|---|---|---|---|
| Pleuritis and pleural effusion | ≤20 | Pleurisy, dyspnea | CXR | PE, angina, chest wall disease, infection | NSAIDs, corticosteroids |
| ILD | 20–50 | Often asymptomatic; cough, dyspnea | PFTs, BAL, open-lung biopsy | PAH, aspiration, infection | Corticosteroids with or without CYC |
| PAH | 20–30 | Dyspnea, right heart failure | Echocardiogram, PFTs, BNP | ILD, pulmonary venous hypertension (left heart disease, pulmonary fibrosis), CVTED | Vasodilators and antiproliferative agents (ETRAs, prostanoids, phosphodiesterase type 5 inhibitors) |
| Immunosuppression—corticosteroids with or without CYC | |||||
| Venous thromboembolic disease | Rare | Acute dyspnea, pleurisy, cough, hemoptysis | Spiral CT, ventilation/perfusion scan, D-dimer, antiphospholipid antibody testing | Infection, vasculitis, pleuritis | Anticoagulation |
| Aspiration pneumonitis | Rare | Acute dyspnea with cough, classically right lower lobe infiltrate, fever; can progress to ARDS | CXR, PFTs, BAL, barium swallow | Infection, ILD | Aspiration/reflux therapy, antibiotics |
| Alveolar hemorrhage | Rare | Acute dyspnea, patchy acinar infiltrates with bibasilar predominance, fever, hemoptysis, rapid decline in hemoglobin | CXR, BAL, serial hemoglobin | Infection, PE, Goodpasture syndrome, SNV | Corticosteroids, CYC |
| Pulmonary vasculitis | Rare | Dyspnea, hemoptysis, patchy interstitial or alveolar infiltrates | PFTs, open-lung biopsy | Alveolar hemorrhage, PE, infection, SNV; may be cause of PAH | Corticosteroids, CYC |
| Obstructive airways disease | Rare | Wheezing, cough; nocturnal symptoms may predominate if aspiration is the cause | PFTs, barium swallow | Likely associated with reflux/aspiration from esophageal dysmotility | Aspiration/reflux therapy, bronchodilators, consider corticosteroids |
Inflammatory and fibrosing ILD affects 66% of patients with MCTD. Most patients with abnormally low lung volume or DLCO on PFTs are asymptomatic, although an unknown subset of these patients develop progressive interstitial inflammation and pulmonary fibrosis. The lower rate of progressive pulmonary fibrosis differentiates lung disease in MCTD from ILD in scleroderma, which progresses to marked fibrosis and severe pulmonary functional decline in a greater portion of patients. Patients with MCTD who develop ILD typically present with progressive dyspnea on exertion. Dry cough also is reported. PFTs most commonly show a restrictive pattern with reduced lung volumes and decreases in DLCO. PFTs are considered to be more sensitive technology for detecting ILD. Characteristic high-resolution CT scan findings include septal thickening and ground-glass opacities with peripheral and basilar lung zone predominance. Traction bronchiectasis and honeycomb changes form later in the disease process, typically with a basilar, subpleural distribution. As in scleroderma-associated ILD, rare cases of ILD associated with MCTD may manifest with dyspnea and abnormal PFTs, but normal-appearing chest x-ray. BAL may be a useful diagnostic tool in this setting. Most patients with ILD show a neutrophilic or eosinophilic predominance in the cell count differential.
Treatment of ILD in patients with MCTD depends on the severity of the process at the time of diagnosis. Most cases seem to be mild, so corticosteroids are the initial immunosuppressant of choice. In patients without symptomatic improvement and decreased lung volumes and DLCO on follow-up PFTs (within 3 months), the addition of cyclophosphamide should be considered. Mostly small, retrospective reports and case studies seem to support the efficacy of cyclophosphamide. In patients presenting with significant pulmonary functional loss, substantial fibrosis, and intensely inflammatory BAL fluid, or a rapidly progressing course, the clinician should consider combination therapy with corticosteroids and cyclophosphamide at the outset of treatment.
Our center uses the same treatment regimen in scleroderma-associated ILD and moderate to severe ILD associated with MCTD, which incorporates intravenous pulses of cyclophosphamide (0.5 to 1 mg/kg/mo) with tapering doses of corticosteroids. The most appropriate duration of cyclophosphamide therapy is unknown at this time for ILD associated with MCTD. Our center administers 6 to 12 monthly doses depending on the severity of the lung disease and the quality of the initial clinical response. Corticosteroids may induce scleroderma-renal crisis; their use in scleroderma lung disease should be minimized or used cautiously. Because of the low risk of scleroderma renal crisis in MCTD, initial corticosteroid doses are more aggressive, beginning at 1 mg/kg/day and tapering over 3 to 6 months. Patients should be monitored closely with at least quarterly PFTs and scans at least every 6 months. Serial 6-minute walk tests provide a useful measure of changes in functional capacity. In addition, measuring pulse oximetry during the test can assess the need for supplemental oxygen.
In children, ILD has been reported in mild and severe forms. There is no evidence for a difference in the prevalence or clinical characteristics of ILD in children compared with adults with ILD. Because ILD in this disease tends toward pulmonary fibrosis, it is important to monitor all patients for ILD with frequent PFTs, preferably every 3 to 6 months in the first few years after diagnosis, and then annually. Patients who develop dyspnea and restrictive pattern on PFTs should undergo or experience a decrease in DLCO. If the result is unremarkable, and no other explanation is available for the pulmonary decline, BAL is indicated to confirm the presence of interstitial inflammation. In these cases, BAL also may be a useful outcome measure, especially if the scan fails to show changes.
The leading cause of death in MCTD is PAH. PAH is now recognized to develop in nearly one third of MCTD patients and has been reported in children and adults. Postmortem studies have shown intimal proliferation and medial smooth muscle hypertrophy in the small vessels of the lungs, occasionally associated with superimposed thrombosis and plexiform lesions. The histologic features in MCTD are identical to the features observed in PAH patients with scleroderma. Because antiphospholipid antibody syndrome is far less common in patients with MCTD, PAH developing as a consequence of chronic venous thromboembolic disease is unlikely. In all patients who develop PAH, routine screening for underlying causes of secondary PAH should be done, including screening for chronic venous thromboembolic disease, HIV, sickle cell disease, and congenital heart disease. The natural history of PAH in MCTD is unclear. Although there are rare reports of remission in patients treated with immunosuppressive and other drugs, progressive PAH followed by right heart failure is the leading cause of death in MCTD. PAH progresses more rapidly in patients with underlying connective tissue disorders than in patients with idiopathic PAH. Annual screening with echocardiogram, PFTs, serum brain natriuretic peptide, and 6-minute walk test is a key component in MCTD management. PAH may develop 20 years after diagnosis.
PAH in patients with MCTD has rarely been reported to improve with immunomodulatory therapy. Because responsiveness to immunosuppression is not characteristic of PAH in scleroderma, it is possible that some patients with MCTD may develop PAH as a consequence of an immune-mediated inflammatory process, such as a vasculitis. To date, there are no substantiating pathologic data to support this hypothesis. The rate of response to immunosuppressive therapy suggests, however, that treatment of PAH in patients with MCTD should incorporate a course of corticosteroids and intravenous pulse cyclophosphamide for 3 to 6 months. In our center, all patients with MCTD are concomitantly treated with standard vasoactive medications, such as endothelin receptor antagonists or phosphodiesterase type 5 inhibitors. We may initiate parenteral prostanoid therapy (continuous subcutaneous treprostinil or continuous intravenous epoprostenol) in patients with rapidly progressing PAH, fulminant or severe right heart failure, or extremely poor functional status. At present, there is no clear evidence of efficacy or safety favoring any single PAH treatment in MCTD.
Sjögren Syndrome
Sjögren syndrome is a chronic autoimmune disease that primarily affects the lacrimal and salivary glands. The cardinal clinical features are parotitis, keratoconjunctivitis sicca, and xerostomia; there is variable systemic involvement. Clinical manifestations include dry eyes, dry mouth with oral ulcerations, dental caries, salivary gland swelling, difficulty swallowing, vulvovaginitis, and gastrointestinal problems. The hallmark autoantibody findings include a positive ANA, Ro (SS-A), La (SS-B), and rheumatoid factor (RF). Additionally, patients with Sjögren syndrome often have an elevated erythrocyte sedimentation rate and hypergammaglobulinemia. Similar to other autoimmune diseases, it affects females significantly more often than males, with a ratio of approximately 9:1. Sjögren syndrome may occur alone (primary) or in association with other connective tissue diseases, such as rheumatoid arthritis, SLE, and systemic sclerosis (secondary).
In the pediatric population, Sjögren syndrome is extraordinarily rare with a mean age of onset of 10 years. A review of 145 pediatric cases revealed parotid enlargement in 70%, eye involvement in 66%, and xerostomia in 43%. A positive ANA was detected in 78%. Antibodies to Ro, La, and RF were positive in 74%, 65%, and 66% of patients. Depending on which modality is used, pulmonary involvement in adult Sjögren syndrome has been estimated at 9% to 75%. In contrast to adults, pulmonary involvement is rare in children.
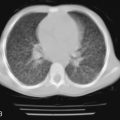
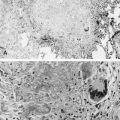
Pulmonary involvement can manifest clinically with progressive dyspnea and dry cough. Physical examination may reveal bibasilar crackles; digital clubbing is usually absent. PFTs typically reveal a restrictive pattern with diminished DLCO. Chest radiographs are less sensitive than high-resolution CT, but may show reticular or nodular opacities. Findings include ground-glass opacities, honeycombing, lung cysts, consolidation, and bronchiectasis. A CT scan from a patient with Sjögren syndrome is shown in Figure 10-3 . Although BAL is not routinely warranted, it may assist in excluding an infectious etiology. BAL can show evidence of lymphocytic or neutrophilic alveolitis, the latter associated with more progressive disease. Various histopathologic patterns are seen, including lymphocytic interstitial pneumonia, nonspecific interstitial pneumonia, usual interstitial pneumonia, cryptogenic organizing pneumonia, primary pulmonary lymphoma, and fibrosis. The hallmark finding on lung histopathology is a CD4 + polyclonal lymphocytic and plasma cell infiltrate. Figure 10-4 shows pulmonary histopathology from a patient with Sjögren syndrome who shows a lymphocytic interstitial pneumonia pattern.


Treatment of ILD in Sjögren syndrome centers on immunosuppression, usually with systemic corticosteroids. No controlled trials have examined the efficacy of alternative immunosuppressive agents, such as azathioprine, cyclosporine, and cyclophosphamide. Treatment needs to be adjusted depending on the severity of disease and the presence or absence of other systemic features.
Juvenile Rheumatoid Arthritis
Juvenile rheumatoid arthritis (JRA) is a chronic inflammatory disease of unclear etiology, which can manifest with articular and extra-articular involvement. The American College of Rheumatology (ACR) has classified JRA into three subtypes : (1) polyarthritis characterized by involvement of five or more inflamed joints, (2) oligoarthritis with involvement of fewer than five inflamed joints, and (3) systemic onset with features of arthritis and characteristic fever. Systemic-onset JRA is most commonly associated with extra-articular manifestations, but the other subtypes may be complicated by nonarticular organ involvement. Lung involvement, although uncommon, can have deleterious consequences when not recognized and treated.
The pleuropulmonary manifestations of JRA can be classified as involvement of the parenchyma, airways, pleura, and pulmonary vasculature. These manifestations include parenchymal or pleural infection, interstitial pneumonia/pneumonitis (IP), interstitial fibrosis, BOOP, rheumatoid nodules, bronchiectasis, chronic obstruction, obstructive bronchiolitis, pleuritis, pleurisy, pulmonary vasculitis, and PAH. When referring to the involvement of lung parenchyma, the term “interstitial lung disease” has been applied. The pulmonary toxicities of medications used to treat JRA (discussed subsequently) can add a level of complexity when assessing the impact of the disease.
In contrast to adults with rheumatoid arthritis, lung involvement in JRA historically has been seen with much less frequency. Pleuropulmonary involvement has been reported in 1% to 40% of adults with rheumatoid arthritis. This range is influenced by the method used to detect evidence of lung disease. Earlier studies relied on data obtained from chest x-rays and PFTs. The use of high-resolution CT of the chest has improved our ability to identify lung disease. In a prospective assessment of 150 rheumatoid arthritis patients, 20% had findings on CT consistent with ILD. Data on the prevalence of pleuropulmonary involvement in children with JRA are sparse. A study of 191 patients with JRA at a single academic center identified evidence of lung or pleural involvement in 4% of children. Most of the children (75%) had systemic-onset JRA. A smaller study examined the prevalence of PFT abnormalities in patients with JRA and found asymptomatic abnormal PFTs in 60%, half of whom had subnormal DLCO. Only one patient had an abnormal chest radiograph. In contrast, PFT measurements in a separate population of JRA patients did not identify any differences between DLCO, total lung capacity (TLC), or FEV 1 compared with controls. Children with JRA had lower FVC and peak expiratory flow rates, which were attributed to respiratory muscle weakness rather than intrinsic lung disease.
Pleuropulmonary manifestations of JRA can manifest indolently and can precede the onset of arthritis. Data, particularly those obtained from PFTs, suggest that there may be a prodromal period of asymptomatic disease. No longitudinal studies have assessed the significance of abnormal PFT measurements in children with JRA. The available data suggest that the prevalence of lung disease remains low in these patients.
In circumstances where pleuropulmonary involvement precedes onset of arthritis, patients can present with cough, dyspnea, and tachypnea. A prior history of recurrent pulmonary infiltrates, often treated as infections, has been reported. Patients with a known diagnosis of JRA can present with similar subjective symptoms of cough, dyspnea, and tachypnea in addition to chest pain. Physical examination findings may reveal digital clubbing, cyanosis, bibasilar crackles on chest auscultation, nasal flaring, tachycardia, tachypnea, and hypoxia. The presence of clubbing is often an indicator of prolonged disease.
Occasionally, the objective physical examination findings can be difficult to interpret in the setting of active arthritis, especially in patients with systemic-onset JRA. These patients can have a febrile illness associated with arthritis, rash, lymphadenopathy, hepatosplenomegaly, elevated inflammatory markers erythrocyte sedimentation rate, and C-reactive protein, leukocytosis, and serositis. Under these circumstances, it is important to be vigilant for other mimics of chronic inflammatory arthritis, such as infection and malignancy. If the physical examination suggests a pleuropulmonary process, it is advisable to assess the problem thoroughly.
The use of PFTs for diagnosing and screening for pleuropulmonary disease in JRA is of paramount importance. A diminished DLCO has been observed in 0% to 30% of children with JRA. In controlled studies, significant decreases of FVC and peak expiratory flow have been observed, but were attributed to respiratory muscle weakness. Most PFT abnormalities can be described as restrictive lung defects with impaired DLCO. An isolated depression of DLCO can be seen independently of restrictive or obstructive lung physiology. The lack of longitudinal PFT assessment studies in JRA has limited the ability to interpret these findings. Several case reports have documented improvement or stabilization of PFTs in children with JRA over time, many of whom were treated with immunosuppressive therapy. Progressively worsening PFTs portend greater morbidity and mortality.
An additional use of PFTs is to focus on the assessment of PAH. Although uncommon in patients with JRA, primary PAH has been seen in a child with systemic-onset JRA. In this patient, all parameters of pulmonary function were normal except for DLCO, which was 35% of normal. The authors have seen two patients whose course of systemic-onset JRA was complicated by PAH. Both patients died as a result of their disease. Although PFTs may suggest PAH, the diagnosis is confirmed by direct measurement of pulmonary arterial pressure by a right heart catheterization.
Chest radiographs have frequently failed to provide important qualitative descriptions or indications of disease severity in rheumatoid arthritis. Of 28 adults with rheumatoid arthritis with evidence of ILD, only 36% had abnormal chest radiographs. Multiple case reports of children with active pleuropulmonary manifestations of JRA showed objective abnormal findings on chest radiographs, however. Using a chest radiograph to screen for JRA in an asymptomatic child may be of limited clinical utility.
Findings include pleural effusions, patchy pulmonary infiltrates, reticulonodular infiltrates, and alveolar infiltrates with air bronchograms. One patient presented with a pneumomediastinum in the setting of organizing bronchiolitis. Other findings include ground-glass opacities, reticular lesions, honeycombing, intralobular septal thickening, bronchiectasis, consolidations, pleural effusions, pleural irregularities, and mediastinal lymphadenopathy.
Studies using high-resolution CT scan of the chest have shown the greater sensitivity of this diagnostic study compared with conventional chest radiographs in detecting ILD. High-resolution CT provides the opportunity to detect ILD at an earlier stage and potentially to improve long-term prognosis.
The pleuropulmonary manifestations of JRA can be categorized as manifestations involving the pleura, parenchyma, airways, or vasculature. In contrast to adults with rheumatoid arthritis–related ILD, pulmonary rheumatoid nodules are rare in children with JRA. Rheumatoid nodules, which are usually present at the elbows and distally, are classically seen in patients who are RF-positive. Histologic examination of a rheumatoid nodule is characterized by a central area of fibrinoid necrosis surrounded by palisading epithelioid histiocytes, which is surrounded further by a matrix of lymphocytes, plasma cells, and fibroblasts. Most children with JRA are RF-negative. Population-based estimates have documented positive RF in 3% to 42% of children with JRA. The greater presence of positive RF was seen in an aboriginal Canadian population with a known HLA association with rheumatoid arthritis. Of children who have a positive RF, most are adolescents and have a polyarticular course that mimics adult rheumatoid arthritis.
When feasible, pleural fluid assessment can help identify the underlying cause of pleuritis. Pleural fluid suggestive of an exudative rheumatoid effusion is often characterized as follows: lymphocyte-predominant leukocytosis (100 to 7000 cells/L), low glucose (<50 mg/dL), elevated lactate dehydrogenase (>1000 IU/L), elevated protein (>4 g/dL), and depressed complements. It is necessary to exclude infection because this is also one of the few conditions associated with low pleural fluid glucose levels. In patients with systemic-onset JRA, pleuritis is the most common pleuropulmonary complication.
BAL offers potential advantages in assessing ILD in JRA. It allows for analysis of inflammatory cells, and can aid in excluding infectious etiologies in acute and chronic processes. Compared with open lung biopsy, it is less invasive. Studies in adults with rheumatoid arthritis–related ILD have shown poor correlation between BAL findings and PFTs. There was a trend toward increased numbers of neutrophils in patients with usual interstitial pneumonia compared with nonspecific interstitial pneumonia. Overlap was noted on BAL findings, however, in patients with usual interstitial pneumonia and patients with inflammatory airway disease with organizing pneumonia. These findings highlight the difficulty in interpreting BAL specimens, but do not obviate the utility of BAL to exclude infection.
Lung biopsy remains the gold standard for assessing pleuropulmonary disease in JRA. An appropriate sample can provide information about parenchymal, pleural, airway, and vascular involvement. Although a lung biopsy is not necessary in every JRA patient with an acute pulmonary process, it should be considered in circumstances in which therapy is predicated on discerning between an aseptic inflammatory process, an infectious process, and a vascular process.
Most, but not all, children with JRA with biopsy-proven pleuropulmonary disease are RF-positive. Lung biopsy specimens were obtained in patients who had radiographic evidence of parenchymal disease. In one patient who was diagnosed with primary PAH, a lung biopsy specimen was obtained to exclude vasculitis in the setting of normal imaging studies. The predominance of lung biopsy specimens obtained from patients with parenchymal disease adds an element of selection bias, particularly with regard to assessing pleural disease.
Findings on lung biopsy have shown lymphoid interstitial pneumonitis, IP with lymphoid bronchiolitis, isolated lymphoid bronchiolitis, BOOP, IP with thickened alveolar septa, and septal lymphocytic IP with prominent lymphoid follicles and germinal centers. An adolescent girl with seropositive JRA complicated by small vessel vasculitis developed ILD with evidence of severe pulmonary fibrosis and arteritis of the pulmonary vessels on lung biopsy specimen. The family history was positive for seropositive JRA with ILD in the patient’s mother. One child with systemic-onset JRA who was RF-negative developed pulmonary interstitial and intra-alveolar cholesterol granulomas, also called “lipoid” pneumonia. Whether this pneumonia was related to the underlying disease or therapy was unclear because the patient had been treated with chlorambucil, cyclophosphamide, and methotrexate. A child with seronegative systemic-onset JRA underwent lung biopsy with objective evidence of PAH. Histology showed pulmonary vascular obstruction with findings of intimal fibrosis of the small and medium-sized pulmonary arteries. Postmortem pleuropulmonary evaluation of 16 patients with a known diagnosis of JRA found radiographically and clinically detectable pulmonary lesions in 70%. Gross assessment showed lung edema in 13 patients, pleural effusions in 3 patients, and pulmonary amyloidosis in 3 patients, and no pulmonary nodules. Most of the patients died of complications of amyloidosis, and many had multiorgan failure that may have contributed to the pulmonary findings at autopsy.
The pathogenesis of pleuropulmonary disease in JRA is incompletely understood. Data collected from adults with rheumatoid arthritis suggest aberrant T cell and B cell activation in patients with extra-articular disease. Patients with rheumatoid arthritis with interstitial pneumonitis (usual interstitial pneumonia or nonspecific interstitial pneumonia) have significantly greater levels of CD4 + T cells and, to a lesser extent, CD3 + cells on lung biopsy specimens compared with patients with idiopathic IP, suggesting a possible crucial role of CD4 + T cells in the pathogenesis of rheumatoid arthritis–associated IP. Similarly, patients with rheumatoid arthritis with IP have increased numbers of CD20 + B cells on lung biopsy specimens. Aggregates of CD20 + B cells were found predominantly in a peribronchiolar distribution. The central role of B lymphocytes in the pathogenesis of rheumatoid arthritis has been well described and is supported by the effectiveness of targeted B cell therapy in patients with rheumatoid arthritis. Whether these findings suggest a direct role of B cells in the pathogenesis of rheumatoid arthritis–related IP remains to be determined. In addition, patients with rheumatoid arthritis with IP have greater numbers of mast cells on lung biopsy specimens. It is unclear whether the increased presence of mast cells is secondary to elevated CD4 + T cells or underlying pulmonary fibrosis. It is possible that mast cells or mast cell mediators induce the release of transforming growth factor-β, which is associated with the development of pulmonary fibrosis. The elucidation of these pathways is key to the identification of novel therapeutic targets.
To date, no randomized trials have shown the efficacy or superiority of immunosuppressive agents in treating rheumatoid arthritis–associated ILD. Limited experience in treating children with pleuropulmonary disease and JRA produced variable results. Although some patients had complete resolution of their lung disease, others had chronic or progressive ILD as evidenced objectively and subjectively.
Glucocorticoids have been the mainstay of therapy for pleuropulmonary disease in JRA. Additional immunosuppressive agents that have been used with variable success include cyclophosphamide, cyclosporine, intravenous immunoglobulin, salicylates, parenteral gold, methotrexate, etanercept, penicillamine, and hydroxychloroquine. The evidence does not indicate clearly whether prognosis may correlate with initiation of therapy early in the course of lung disease. When monitored serially, patients with baseline evidence of restrictive lung disease often continue to have a component of restrictive lung function. Complete resolution of radiographic abnormalities might be predictive of a good pulmonary prognosis. When isolated PAH is found with JRA, it should be treated similarly to PAH associated with scleroderma, with emphasis on the use of pulmonary vasodilators used in idiopathic PAH. Even with advances in therapeutics, however, it is important for care providers and families to recognize that the pleuropulmonary manifestations of JRA can be fatal despite aggressive immunosuppressive therapy.
Related posts:
 Chronic Lung Disease of Infancy
Chronic Lung Disease of Infancy
 Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
 Pulmonary Manifestations of Endocrine and Metabolic Diseases
Pulmonary Manifestations of Endocrine and Metabolic Diseases
 Pulmonary Manifestations of Gastrointestinal Diseases
Pulmonary Manifestations of Gastrointestinal Diseases
 Pulmonary Manifestations of Systemic Vasculitis
Pulmonary Manifestations of Systemic Vasculitis
 Pulmonary Manifestations of Dermatologic Diseases
Pulmonary Manifestations of Dermatologic Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


