Pulmonary Vascular Disease
Curtis E. Green
Jeffrey S. Klein
Pulmonary Edema
Basic Principles. Under normal conditions, the interstitial space of the lung is kept dry by pulmonary lymphatics located within the axial and peripheral interstitium of the lung. The lymphatics drain the small amounts of transudated fluid that enters the interstitial spaces as an ultrafiltrate of plasma. Because there are no lymphatic structures immediately within the alveolar walls (parenchymal interstitium), filtered interstitial fluid is drawn to the lymphatics by a pressure gradient from the alveolar interstitium to the axial and peripheral interstitium. When the rate of fluid accumulation in the interstitium exceeds the lymphatic drainage capabilities of the lung, fluid accumulates first within the interstitial space. As the amount of extravascular fluid increases, fluid accumulates in the corners of the alveolar spaces. Progressive fluid accumulation eventually produces flooding of the alveolar spaces, resulting in airspace pulmonary edema. While interstitial edema may leave the gas-exchanging properties of the lung unaffected, flooding of the alveolar spaces leads to impaired oxygen and carbon dioxide exchange.
Excess fluid accumulation in the lung is caused by one of three basic mechanisms. The most common mechanism involves a change in the normal Starling forces that govern fluid movement in the lung. Because normal fluid movement is determined by the differences in hydrostatic and oncotic pressure between the pulmonary capillaries and surrounding alveolar interstitium, an imbalance in these forces may lead to pulmonary edema. This imbalance of forces is most commonly the result of increased capillary hydrostatic pressure (hydrostatic pulmonary edema) and less commonly diminished plasma oncotic or interstitial hydrostatic pressure. A second mechanism is obstruction or absence of the normal pulmonary lymphatics, which leads to the excess accumulation of interstitial fluid. Thirdly, a wide variety of disorders can injure the epithelium of the capillaries and alveoli, causing an increase in capillary permeability that allows protein-rich fluid to escape from the capillaries into the pulmonary interstitium.
Imaging findings in pulmonary edema result from both the interstitial and the airspace components and depend to some extent on the cause of the edema, as will be discussed later. The radiographic appearance of interstitial pulmonary edema results from thickening of the components of the interstitial spaces by fluid (1). Thickening of the axial interstitium results in the loss of definition of the intrapulmonary vascular shadows and thickening of the peribronchovascular interstitium causing peribronchial cuffing and tram tracking. Edema within alveolar septa is not discernible as discrete opacities but produces ground-glass opacity initially in only the dependent lung zones and then throughout the lungs, but still worse in the dependent zones. Involvement of peripheral and subpleural interstitial structures produces Kerley lines and subpleural edema. Kerley A and B lines represent thickening of central connective tissue septa and peripheral interlobular septa, respectively, whereas Kerley C lines represent a network of thickened interlobular septa (Fig. 14.1). Subpleural edema is the accumulation of fluid within the innermost (interstitial) layer of the visceral pleura and is best seen on the lateral radiograph as smooth thickening of the interlobar fissures. The radiographic changes of interstitial pulmonary edema may progress to those of airspace edema or, if successfully treated, resolve within 12 to 24 hours.
Airspace pulmonary edema develops when fluid in the interstitial spaces spills into the alveoli. The upright chest radiograph typically shows bilaterally symmetric airspace opacities predominately in the mid and lower lung zones. Airspace nodules and the findings of interstitial edema (Kerley B lines and subpleural edema) are usually present peripherally. As with interstitial edema, the airspace opacities of alveolar edema may change rapidly, often within hours. The differential diagnosis of diffuse airspace opacities has been reviewed (see Table 12.9).
Thin-section CT can on occasion be quite useful for identification and assessment of pulmonary edema as the findings are fairly specific (2). Thickening of subpleural, septal, and bronchovascular structures is well depicted. Mild parenchymal edema produces a ground-glass pattern around the hila (Fig. 14.2). Early alveolar edema is seen as centrilobular airspace nodules surrounding the arteries within the lobular core, whereas severe alveolar edema produces dense perihilar airspace opacification.
Hydrostatic pulmonary edema (normal capillary permeability) is the most common form of pulmonary edema. It is usually caused by an elevation in the pulmonary venous pressure (pulmonary venous hypertension [PVH]). The classic cause of PVH is left ventricular systolic failure, but renal failure and a variety of cardiac and noncardiac abnormalities have the same physiology. Decreased capillary oncotic pressure, such as present in patients with hypoalbuminemia secondary to the nephrotic syndrome or liver failure, can cause findings identical to those in patients with elevated hydrostatic pressure.
The causes of PVH may be divided into four major categories: obstruction to left ventricular inflow, left ventricular systolic dysfunction (LV failure), mitral valve regurgitation, and systemic or pulmonary volume overload. The classic cause of obstruction to left ventricular inflow is mitral stenosis, but
poor left ventricular compliance (diastolic dysfunction), such as caused by hypertrophy or chronic ischemic subendocardial fibrosis, is more common. Mimickers of mitral stenosis such as left atrial myxomas are rare. Obstruction of the central pulmonary veins from tumor, fibrosing mediastinitis, or pulmonary vein thrombosis may also be associated with the radiographic findings of PVH. Common causes of LV failure include ischemic heart disease, aortic valve stenosis and regurgitation, and nonischemic cardiomyopathy (Table 14.1). Severe mitral valve regurgitation can cause PVH directly by elevating left atrial pressure or secondarily by causing LV failure. Acute pulmonary volume overload is relatively common and most frequently due to iatrogenic overhydration. Acute postinfarction ventricular septal defect is a rare cause. Patients with acute or chronic renal failure may develop pulmonary edema because of increased pulmonary capillary hydrostatic
pressure caused by a combination of hypervolemia and LV dysfunction.
poor left ventricular compliance (diastolic dysfunction), such as caused by hypertrophy or chronic ischemic subendocardial fibrosis, is more common. Mimickers of mitral stenosis such as left atrial myxomas are rare. Obstruction of the central pulmonary veins from tumor, fibrosing mediastinitis, or pulmonary vein thrombosis may also be associated with the radiographic findings of PVH. Common causes of LV failure include ischemic heart disease, aortic valve stenosis and regurgitation, and nonischemic cardiomyopathy (Table 14.1). Severe mitral valve regurgitation can cause PVH directly by elevating left atrial pressure or secondarily by causing LV failure. Acute pulmonary volume overload is relatively common and most frequently due to iatrogenic overhydration. Acute postinfarction ventricular septal defect is a rare cause. Patients with acute or chronic renal failure may develop pulmonary edema because of increased pulmonary capillary hydrostatic
pressure caused by a combination of hypervolemia and LV dysfunction.
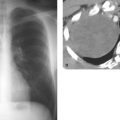
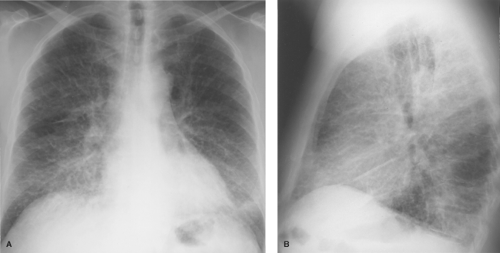 Figure 14.1. Interstitial Pulmonary Edema Caused by Cardiac Disease. Posteroanterior (A) and lateral (B) chest films in a 65-year-old man with an anterior wall myocardial infarction show hydrostatic interstitial edema as evidenced by prominent upper lobe vessels (redistribution of pulmonary blood flow), indistinct lower lung zone pulmonary vessels, peripheral linear opacities (thickened interlobular septa or Kerley B lines), and thickened fissures (subpleural edema) resulting from acute left ventricular failure. Note the absence of cardiomegaly. |
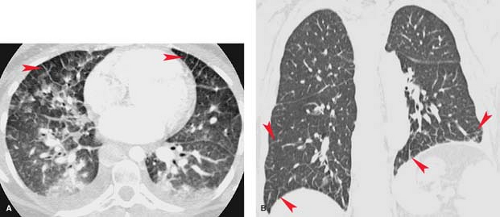 Figure 14.2. Thin-Section CT of Hydrostatic Interstitial Pulmonary Edema. A. Axial scan in a 28-year-old woman with postpartum cardiomyopathy shows thickening of the interlobular septal (arrowheads) and bronchovascular bundles with dependent patchy ground-glass and airspace opacities and bilateral pleural effusions. B. Coronal reconstruction in a different patient with hydrostatic interstitial edema. Thickened septal lines (arrowheads) and bronchovascular bundles and scattered ground-glass opacities are present, but there is no airspace consolidation. |
Table 14.1 Causes of Pulmonary Venous Hypertension and Pulmonary Edema | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The classic radiographic findings of PVH are enlargement of pulmonary veins and redistribution of pulmonary blood flow to the nondependent lung zones (1). Pulmonary venous enlargement is seen as progressive dilatation of horizontally oriented pulmonary veins on serial chest radiographs. The redistribution of pulmonary blood flow results from lower zone pulmonary venous constriction causing increased resistance to lower zone blood flow, with resultant preferential flow into upper lobe vessels. Therefore, with PVH in the upright patient with normal lung parenchyma, the upper zone vessels are frequently as large as or larger in diameter than the lower zone vessels. This is the opposite of the normal appearance, in which the lower zone vessels are larger than the upper zone vessels as a result of the normal gravitational effects on pulmonary blood flow. It should be noted that in patients with basilar lung disease, pulmonary blood flow may appear to be redistributed in the absence of PVH and with upper lobe lung disease (e.g., centrilobular emphysema) distribution may not change.
The sequence of events following the development of PVH has been studied in patients with acute cardiac decompensation following myocardial infarction. Several studies have correlated the radiographic findings of PVH in the erect patient with measurements of pulmonary capillary wedge pressure (PCWP) using flow-directed balloon occlusion (e.g., Swan–Ganz) catheters. When PCWP is normal (8 to 12 mm Hg), the chest radiograph is normal. Mild elevation of PCWP (12 to 18 mm Hg) produces constriction of lower lobe vessels and enlargement of upper lobe vessels. Progressive elevation of PCWP (19 to 25 mm Hg) leads to the findings of interstitial pulmonary edema: loss of vascular definition, peribronchial cuffing, and Kerley lines (Fig. 14.1). PCWP above 25 mm Hg produces alveolar filling with radiographic findings of bilateral airspace opacities in the perihilar and lower lung zones.
Atypical Radiographic Appearances of PVH. Several conditions may give rise to atypical radiographic appearances of PVH. Because the distribution of edema is affected by gravity, it is not surprising that edema fluid accumulates posteriorly or unilaterally in patients maintaining a prolonged supine or decubitus position, respectively. The diagnosis of unilateral edema is suggested by typical radiographic and clinical findings of pulmonary edema in one lung that resolve rapidly or redistribute with changes in patient positioning. Another cause of asymmetric or unilateral pulmonary edema is an interruption in the blood supply to one lung. This may be seen in pulmonary artery hypoplasia or in an acquired obstruction to pulmonary arterial blood flow, such as central pulmonary embolus or extrinsic compression of the pulmonary artery from tumor or fibrosis. In these conditions, the lung with diminished pulmonary blood flow is “protected” from the transudation of fluid and the development of pulmonary edema. Bronchogenic carcinoma, lymphoma, or other causes of unilateral lymph node enlargement can impede normal lymphatic drainage and predispose to unilateral pulmonary edema. Similarly, unilateral pulmonary venous obstruction from tumor or fibrosing mediastinitis will predispose to edema on the affected side. Unilateral pulmonary edema may develop in the lung that is reexpanded by the rapid evacuation of a large pleural fluid collection or pneumothorax. This is known as reexpansion pulmonary edema and is discussed in a subsequent section.
Alveolar pulmonary edema localized to the right upper lung may be seen in patients with severe mitral regurgitation. Edema formation is likely the result of preferential regurgitant flow of blood into the right upper lobe pulmonary vein across the superiorly and posteriorly oriented mitral valve. These patients will usually have typical radiographic findings of interstitial edema elsewhere in the lungs.
Patients with pulmonary emphysema frequently have unusual appearances of alveolar edema. Areas of bullae, most commonly in the apical portions of the lungs, are spared from the development of alveolar edema because the pulmonary blood flow to these regions has already been obliterated by the emphysematous process. These emphysematous regions within adjacent areas of airspace opacification can simulate cavity formation and may be difficult to distinguish radiographically from necrotizing pneumonia or pneumatocele formation. Comparison with previous radiographs and correlation with the clinical course will aid in the proper diagnosis.
Increased Capillary Permeability Edema. Rapidly progressive respiratory compromise caused by leakage of protein-rich edema fluid into the lung, resulting from damage to the pulmonary microcirculation, may develop as a complication of a variety of systemic conditions. When respiratory failure develops as a result of this condition and is associated with increased lung stiffness (noncompliance) it is termed acute respiratory distress syndrome (ARDS) (3). The edema associated with this syndrome is called lung injury or increased capillary permeability edema, as compared to the normal alveolocapillary permeability of hydrostatic edema. Many pulmonary and nonpulmonary disorders have been associated with increased-permeability edema (Table 14.2); the most common are shock, severe trauma, burns, sepsis, narcotic overdose, and pancreatitis. Although the precise pathogenesis of capillary permeability edema has yet to be completely elucidated, current evidence suggests that recruitment and activation of neutrophils in the lung with release of enzymes and oxygen radicals are key factors in the development of capillary endothelial damage.
Table 14.2 Etiologies of Increased Permeability Pulmonary Edema | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
The pathologic changes associated with ARDS are those of diffuse alveolar damage and are common to all patients regardless of the underlying etiology. Within 12 to 24 hours following the initial insult (stage 1 ARDS), damage to capillary endothelium produces engorged capillaries and proteinaceous interstitial edema. Within the first week (stage 2), the injury to type 1 pneumocytes leads to the flooding of alveoli with edema fluid and proteinaceous and cellular debris, which form hyaline membranes lining the distal airways and alveoli. In stage 3 ARDS, type 2 pneumocytes proliferate in an attempt to reline the denuded alveolar surfaces, and fibroblastic tissue proliferates within the airspaces. This fibroblastic tissue may resolve and leave minimal scarring or, particularly in those with severe disease and long-standing oxygen requirements, result in extensive interstitial fibrosis.
Radiographically, ARDS follows a predictable pattern. Chest radiographs become abnormal by 12 to 24 hours following the onset of dyspnea and demonstrate patchy peripheral airspace opacities (Fig. 14.3) (2). CT scans show diffuse ground-glass and airspace opacities which may have a striking nondependent distribution. Interlobular septal thickening is usually absent (Fig. 14.4). These opacities coalesce over the next several days to produce confluent bilateral airspace opacities with air bronchograms. Radiographic improvement in the opacities may be seen within the first week, but this is often caused by the effects of increasing positive pressure ventilation rather than true histologic improvement. After 1 week, the airspace opacities gradually give way to a coarse reticulonodular pattern that may resolve over the course of several months or remain unchanged, in which case the pattern represents irreversible pulmonary fibrosis (i.e., honeycombing). Pneumonia complicating ARDS is difficult to diagnose radiographically, but it should be suspected when a focal area of airspace opacification or a significant pleural effusion develops during the course of the disease. Likewise, the superimposition of LV failure may be impossible to recognize but is suggested by rapid clinical and radiographic deterioration associated with changes in measured PCWP and edema fluid protein content. Pneumomediastinum and pneumothorax may result as a complication of positive pressure ventilation to stiff lungs and should be sought on portable chest radiographs.
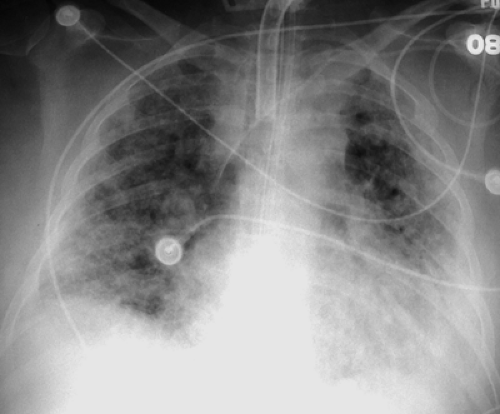 Figure 14.3. Increased Permeability (Lung Injury) Edema in Acute Respiratory Distress Syndrome. Portable chest radiograph in a 46-year-old woman with severe pancreatitis and respiratory failure reveals bilateral airspace opacification with a somewhat peripheral distribution, representing diffuse alveolar damage and permeability edema. |
Radiographic Distinction of Hydrostatic From Increased Capillary Permeability Edema. Beyond identifying the presence of pulmonary edema, the ability to distinguish between types of pulmonary edema has significant diagnostic and therapeutic importance. Measurements of PCWP and transbronchial sampling of pulmonary edema fluid are techniques that accurately distinguish hydrostatic from increased capillary permeability edema. In hydrostatic edema, PCWP measurements are elevated and a protein-poor transudative edema
fluid is present, whereas in increased-permeability edema, there is a normal PCWP and proteinaceous edema fluid is seen. Milne and colleagues have described the chest radiographic findings that can be used to distinguish cardiac and overhydration edema from increased capillary permeability edema (3). In pulmonary edema associated with chronic cardiac failure, the heart is usually enlarged and displays an inverted (redistributed) pulmonary blood flow pattern. The distribution of edema is even from central to peripheral over the lower lung zones. The vascular pedicle, which represents the mediastinal width at the level of the superior vena cava and left subclavian artery, is widened (>53 mm on posteroanterior radiograph), reflecting increased circulating blood volume. Lung volumes are diminished because of decreased pulmonary compliance from edema. Peribronchial cuffing, Kerley lines, and pleural effusions represent interstitial and intrapleural transudation of fluid, respectively. These findings may be difficult to interpret, however. Furthermore, cardiac size per se is not particularly useful in distinguishing cardiac-related edema from other causes of hydrostatic and capillary leak edema for the following reasons: many patients with heart failure will not have radiographically evident cardiac enlargement; many patients with cardiac enlargement are not in failure; and enlargement of the cardiac silhouette may be caused by pericardial fluid, mediastinal fat, and poor lung expansion. Cardiomegaly is best considered evidence of a chronic condition rather than an indicator of a specific problem.
fluid is present, whereas in increased-permeability edema, there is a normal PCWP and proteinaceous edema fluid is seen. Milne and colleagues have described the chest radiographic findings that can be used to distinguish cardiac and overhydration edema from increased capillary permeability edema (3). In pulmonary edema associated with chronic cardiac failure, the heart is usually enlarged and displays an inverted (redistributed) pulmonary blood flow pattern. The distribution of edema is even from central to peripheral over the lower lung zones. The vascular pedicle, which represents the mediastinal width at the level of the superior vena cava and left subclavian artery, is widened (>53 mm on posteroanterior radiograph), reflecting increased circulating blood volume. Lung volumes are diminished because of decreased pulmonary compliance from edema. Peribronchial cuffing, Kerley lines, and pleural effusions represent interstitial and intrapleural transudation of fluid, respectively. These findings may be difficult to interpret, however. Furthermore, cardiac size per se is not particularly useful in distinguishing cardiac-related edema from other causes of hydrostatic and capillary leak edema for the following reasons: many patients with heart failure will not have radiographically evident cardiac enlargement; many patients with cardiac enlargement are not in failure; and enlargement of the cardiac silhouette may be caused by pericardial fluid, mediastinal fat, and poor lung expansion. Cardiomegaly is best considered evidence of a chronic condition rather than an indicator of a specific problem.
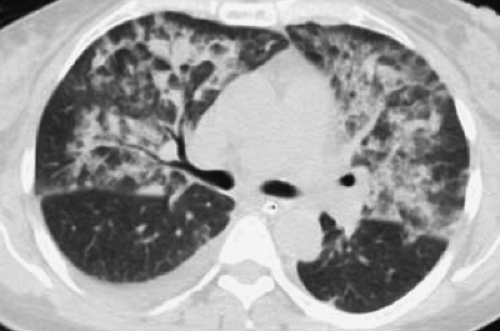 Figure 14.4. Thin-Section CT of Lung Injury Edema. Geographic, nondependent ground-glass and airspace opacities are present, but interlobular septal thickening is absent. |
Capillary permeability edema can sometimes be distinguished from hydrostatic edema by the following: a nondependent or peripheral distribution of edema, an absence of other signs of hydrostatic edema such as interlobular septal thickening and subpleural edema, and, most importantly, a lack of short-term change. It should be noted that some factors may render radiographic distinction of types of pulmonary edema difficult. Radiographs of supine patients will make evaluation of pulmonary blood flow distribution and vascular pedicle width difficult. The presence of severe alveolar edema will obscure underlying vascular markings. Many patients with capillary permeability edema will be overhydrated in attempts to maintain circulating blood volume, producing complex radiographic findings. Lastly, most intubated patients will suffer from more than one problem.
Neurogenic pulmonary edema following head trauma, seizure, or increased intracranial pressure is a complex phenomenon that appears to involve both hydrostatic and increased permeability mechanisms. Massive sympathetic discharge from the brain in these conditions produces systemic vasoconstriction and increased venous return, with resultant increase in LV diastolic pressure and hydrostatic pulmonary edema. The presence of protein-rich edema fluid and normal PCWP in some patients suggests that increased permeability may be a contributing factor.
High-altitude pulmonary edema develops in certain individuals after rapid ascent to altitudes above 3500 m. Edema typically develops within 48 to 72 hours of ascent and appears to reflect a varied individual response to hypoxemia, in which scattered areas of pulmonary arterial spasm result in transient pulmonary arterial hypertension (PAH). This produces an increase in high-pressure blood flow to uninvolved areas, resulting in damage to the capillary endothelium and increased permeability edema, typically with a patchy distribution. Resolution usually occurs within 24 to 48 hours after the administration of supplemental oxygen or a return to sea level.
Reexpansion Pulmonary Edema. Rapid reexpansion of a lung following severe pneumothorax or collapse from a large pleural effusion present more than 48 hours may result in the development of unilateral pulmonary edema. Marked increases in negative pleural pressure following pleural tube placement, impaired pulmonary lymphatic drainage following prolonged lung collapse, and ischemia-induced surfactant deficiency resulting in the need for high negative pleural pressure to reexpand the collapsed lung are proposed mechanisms. Recent evidence points toward prolonged collapse producing ischemia and hypoxemia within the lung, which promotes anaerobic metabolism and formation of free radicals. Reperfusion of the lung upon reexpansion then leads to lung injury and permeability edema. Gradual reexpansion of the lung by slow removal of pleural air or fluid over a 24- to 48-hour period and administration of supplemental oxygen help limit the incidence and severity of this complication.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


